Leucemia de linfocitos grandes granulares de células NK. Reporte de caso
NK-large granular lymphocytic leukemia. A case report
Lucas Condori MK; Manciola FL; Riva ME
Servicio de Hematología, Hospital Interzonal General de Agudos General San Martín de La Plata, Buenos Aires, Argentina.
Palabras claves: leucemia de linfocitos grandes granulares,
células NK,
STAT3.
Keywords: large granular lymphocyte leukemia,
NK cells,
STAT3.
Resumen
La leucemia de linfocitos grandes granulares es un trastorno raro, caracterizado por la expansión clonal de linfocitos con fenotipo de células T (CD3+) en aproximadamente el 85%, mientras que el restante corresponde a células NK (CD3-). Aunque su etiología es desconocida, se ha relacionado con una estimulación antigénica crónica. La presentación clínica puede incluir neutropenia, anemia, esplenomegalia e infecciones recurrentes. El diagnóstico se basa en la identificación de linfocitos grandes granulares atípicos clonales en sangre con la exclusión de expansiones reactivas y una clínica compatible. La inmunosupresión es la terapia de primera línea, siendo efectiva en el 50% en la corrección de citopenias. Se presenta el caso de un paciente masculino de 77 años, sin antecedentes personales relevantes, diagnosticado con leucemia de linfocitos grandes granulares con inmunofenotipo NK y mutación en el gen STAT3, quien inició tratamiento con inmunosupresores y terapia de soporte para citopenias.
Abstract
Large granular lymphocyte leukemia is a rare disorder characterized by the clonal expansion of lymphocytes with a T cell phenotype (CD3+) in approximately 85%, while the remainder corresponds to NK cells (CD3-). Although its etiology is unknown, it has been related to chronic antigenic stimulation. Clinical presentation may include neutropenia, anemia, splenomegaly and recurrent infections. The diagnosis is based on the identification of clonal atypical large granular lymphocytes in the blood with the exclusion of reactive expansions and an in the context of a suitable clinical presentation. Immunosuppression is the first-line therapy, correcting cytopenias in about half of the patients. We present the case of a 77-year-old male patient, with no relevant personal history, diagnosed with large granular lymphocyte leukemia with NK immunophenotype and mutation in the STAT3 gene, who began treatment with immunosuppressants and supportive therapy due to cytopenias.
Introducción
La leucemia de linfocitos granulares grandes (LLGG) es un trastorno linfoproliferativo crónico poco frecuente, definido por una expansión clonal persistente (> 6 meses) de linfocitos con una morfología característica: tamaño grande y núcleos redondeados o dentados y citoplasma abundante con gránulos azurófílos(1).
La LLGG constituye del 2 al 5% de todas las enfermedades linfoproliferativas crónicas en EE. UU y Europa. La incidencia es la misma en hombres y en mujeres y la mediana de edad es 55-60 años(2). Según la quinta clasificación de la Organización Mundial de la Salud (OMS), este trastorno se divide en 2 subtipos según el linaje de las células leucémicas: leucemia de linfocitos granulares grandes T y leucemia de linfocitos granulares grandes NK. (anteriormente denominado trastorno linfoproliferativo crónico de las células NK)(3).
La etiología de LLGG es desconocida. El fenotipo efector/citotóxico terminal de los linfocitos granulares grandes (LGG) sugiere que la enfermedad resulta de una estimulación crónica por antígenos virales, autoantígenos o antígenos tumorales. Las vías de supervivencia de las células como MAPK/ERK, PI3K/AKT, NF-κB, esfingolípidos y JAK/STAT, que conducen a la resistencia a la apoptosis, se encuentran activadas en forma constitutiva(1,6).
En la LLGG la mutación en el gen STAT3 lleva a una activación continua en la vía de señalización JAK-STAT3, que inhibe la apoptosis al aumentar la transcripción de la proteína antiapoptótica Mcl-1. En las células de esta enfermedad se ha descripto también la resistencia a la apoptosis mediada por FAS debido al producto escindido soluble de FAS (sFAS) que interfiere con la unión normal de FAS-L a su receptor.
La detección de mutaciones en STAT3 permiten distinguir las proliferaciones clónales que afectan a las células T y NK de las expansiones reactivas(1,2,4,5).
Un tercio de los pacientes no presentan síntomas al momento del diagnóstico. La presentación clínica inicial incluye neutropenia, que puede presentarse con ulceraciones aftosas orales recurrentes, anemia y esplenomegalia(6).
El diagnóstico de LLGG se basa en la identificación de una población de LGG de células T o NK clonales expandidas de larga duración. Siempre deben excluirse las expansiones de LGG reactivas a infecciones virales o posteriores a la esplenectomía.
El inmunofenotipo en LLGG T más común es CD3+, CD8+, CD57+ y CD16+. Por otro lado, las LLGG NK presentan un perfil caracterizado por CD3−, CD8+, CD16+ y CD56+(1).
El tratamiento está indicado en casos de neutropenia grave (< 0,5×109/L), neutropenia moderada con infecciones recurrentes, anemia dependiente de transfusiones, enfermedad autoinmune asociada y/o esplenomegalia progresiva con aumento rápido de linfocitos sanguíneos circulantes(1,2). Las terapéuticas con metotrexato, ciclosporina, prednisona y ciclofosfamida han demostrado buenos resultados en esta enfermedad. Sin embargo, la resistencia y las respuestas de corta duración representan un desafío continuo, particularmente en aquéllos con anemia refractaria(6,7).
Caso clínico
Paciente masculino de 77 años con antecedentes personales de hipertensión arterial y cardiopatía isquémica. Consultó por un cuadro de astenia, adinamia y pérdida de peso progresiva de 12 meses de evolución. Presentó anemia severa (Hb: 7,1 gr/dL) regenerativa (reticulocitos 10%, índice de producción reticulocitaria: 2.6), recuento leucocitario normal (5.6×109/L) con inversión de la fórmula y neutropenia leve (1.06×109/L). En el extendido de sangre periférica presentó linfocitos de tamaño grande con abundante citoplasma abundante y granulaciones azurófilas (Figura 1).
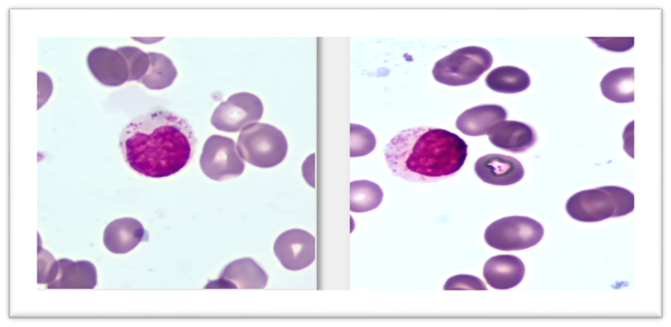
Figura 1. Frotis de sangre periférica de LLGG. Tinción de May Grünwald Giemsa 100x. Linfocito de tamaño grande con un núcleo redondo con cromatina condensada con citoplasma pálido abundante, que contiene una cantidad variable de gránulos azurófilos (policromatofilia y microesferocitos en serie roja por hemólisis).
Fotomicrografía: Lucas Condori MK, Manciola FL, Riva ME. Servicio de Hematología, Hospital San Martín, La Plata, Argentina.
Prueba de Coombs directa (PCD) negativa, bilirrubina total: 2.61 mg% (indirecta 1.93 mg%), LDH: 873 U/L (VN: 100-380), haptoglobina indetectable. Ácido fólico y B12 dentro de parámetros normales. Serologías HIV, VHB, VHC, VHA, VDRL, VEB, HTLV 1-2 no reactivas. TC cuello, tórax, abdomen y pelvis: Esplenomegalia leve (16,4 cm) como único dato positivo.
Con diagnóstico de anemia hemolítica no autoinmune inició tratamiento con prednisona 1 mg/kg/d sin respuesta.
El estudio anatomopatológico de médula ósea mostró normocelularidad con hiperplasia eritroide sin elementos atípicos. La citometría de flujo (CF) de médula ósea no detectó células inmaduras. En CF de sangre periférica se observó un 51% de células de mediano a gran tamaño con complejidad interna escasa con el siguiente fenotipo CD2++, CD7++, CD45++, CD56 - (+ sólo el 6%), CD16+ parcial (76%), CD5-, CD45Ra+, CD45Ro -, HLA DR+/- heterogéneo, CD11c+, CD11a++, Granzima+, Perforina+, CD57-, CD11b -, CD38++, CD4-, CD8 -, CD3cit -. El patrón inmunofenotípico corresponde a células NK. CTG de MO: cariotipo 46,XY [16] y mediante FISH con sonda TCR se observó un patrón de señal normal.
Se realizó estudio por secuenciación de segunda generación (NGS) para búsqueda de mutaciones somáticas en STAT3/STAT5B en el que se detectó la mutación STAT3[G1] .
Inició tratamiento con ciclofosfamida 100 mg/día sin respuesta y persistencia de requerimiento transfusional semanal. Luego de 3 meses de tratamiento se rotó el tratamiento a metotrexato 10 mg/m2 una vez por semana. Ante la persistencia de anemia severa se añadió eritropoyetina 10.000 UI por semana. Mejoró parcialmente los valores hematológicos con disminución del requerimiento transfusional, pero con persistencia de parámetros de hemólisis (Figura 2). En agosto el paciente falleció por shock séptico secundario a SARS-COV2.
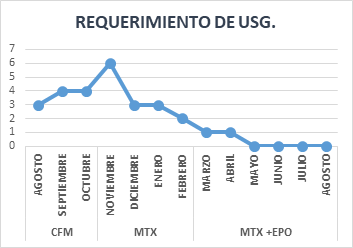
Figura 2. Requerimiento transfusional de unidades de sedimento globular (USG) y la respuesta a inmunosupresores.
Discusión
La LLGG es un trastorno linfoproliferativo crónico infrecuente. El diagnóstico se realiza mediante la clínica, la citometría de flujo y la confirmación de clonalidad. Según el inmunofenotipo se divide en: LLGGT con fenotipo más común CD3+, CD8+, CD57+, CD16+, receptor de células T (RCT) αβ (+), CD4− con o sin expresión de CD56, y LLGG-NK que expresa una expansión CD3-, CD16+, CD56+, con expresión variable de CD57. Para confirmar el diagnóstico se requiere la detección de clonalidad. La clonalidad T debe evaluarse mediante el estudio del rearreglo del receptor de la célula T (TCR) por PCR. En las proliferaciones de células NK el análisis de TCR no tiene utilidad y la clonalidad debe confirmarse mediante el estudio de mutaciones del gen STAT3 o detección de restricción en la expresión de isoformas activadas de los receptores de inmunoglobulinas de células NK (KIRs). Las mutaciones en STAT3, STAT5B y CCL22 son las lesiones genéticas de ganancia de función más comunes identificadas hasta ahora en pacientes con LLGG(3). El estudio citogenético convencional no tiene mayor valor diagnóstico, ya que el cariotipo es normal en la mayoría de los pacientes y no existen anomalías recurrentes. La biopsia ósea no suele aportar información adicional y no está indicada de rutina, pero debe contemplarse en pacientes con citopenias marcadas en los que no se ha podido comprobar clonalidad. Los hallazgos son sutiles y no existe correlación entre el nivel de infiltración medular y la severidad de las citopenias(1,2).
El paciente del caso presentaba inmunofenotipo compatible con proliferación NK y la mutación en STAT3. Los pacientes con mutación en STAT3 tienen mayor frecuencia de neutropenia, anemia, enfermedades autoinmunes y necesidad de tratamiento.
Ante la falta de ensayos prospectivos aleatorios, las opciones de tratamiento estándar actuales en LLGG se basan principalmente en el metaanálisis de ensayos y series de casos. Cuando está indicado, el tratamiento puede realizarse con metotrexato (10 mg/m2 semanal), ciclosporina (1-1,5 mg/kg/ 2 veces al día) o ciclofosfamida (50-100 mg/día). Estos tratamientos son efectivos en casi la mitad de los pacientes, mejorando las citopenias. Nuestro paciente presentó anemia hemolítica no autoinmune con alto requerimiento transfusional y refractaria a los tratamientos inmunosupresores secuenciales utilizados(1,8).
Dentro de la terapia de soporte puede considerarse el uso de eritropoyetina en caso de anemia refractaria con requerimiento transfusional o G-CSF administrado en casos de neutropenia grave(6,7).
Conclusión
La LLGG de clonalidad NK es una entidad muy poco frecuente cuyo diagnóstico se basa en la sospecha clínica y morfológica a partir de la presencia de elementos linfoides granulares característicos. Presenta fenotipo específico (CD3-, CD16+, CD56+) y requiere para la confirmación diagnóstica la detección de clonalidad.
El manejo de la LLGG se basa en terapias inmunosupresoras. Sin embargo, los resultados están lejos de ser óptimos con respuesta globales del 50%, por lo cual es necesario el desarrollo de nuevas terapéuticas.
Conflictos de interés: los autores declaran no poseer conflictos de interés.
Bibliografía
1. Magnano L, Rivero A, Matutes E. Large granular Lymphocytic leukemia: Current state of diagnosis, pathogenesis and treatment. Curr Oncol Rep. 2022;24(5):633–44. Disponible en: http://dx.doi.org/10.1007/s11912-021-01159-y.
2 Rahul E, Ningombam A, Acharya S, Tanwar P, Ranjan A, Chopra A. Large granular lymphocytic leukemia: a brief review. Am J Blood Res. 2022;12(1):17–32.
3. Semenzato G, Calabretto G, Barilà G, Gasparini VR, Teramo A, Zambello R. Not all LGL leukemias are created equal. Blood Rev. 2023;60(101058):101058. Disponible en: http://dx.doi.org/10.1016/j.blre.2023.101058.
4. Zawit M, Bahaj W, Gurnari C, Maciejewski J. Large granular Lymphocytic leukemia: From immunopathogenesis to treatment of refractory disease. Cancers (Basel). 2021;13(17):4418. Disponible en: http://dx.doi.org/10.3390/cancers13174418.
5. Jerez A, Clemente MJ, Makishima H y col. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012;120(15):3048–57. Disponible en: http://dx.doi.org/10.1182/blood-2012-06-435297.
6. Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood. 2017;129(9):1082–94. Disponible en: http://dx.doi.org/10.1182/blood-2016-08-692590.
7. Kwaramba T, Lewis B, Burks B y col. Sustained response to erythropoietin for anemia in NK-cell large granular lymphocytosis: A brief case report. Leuk Res Rep. 2022;17(100292):100292. Disponible en: http://dx.doi.org/10.1016/j.lrr.2022.100292.
8. Lois Iglesias A, Sifuentes Giraldo WA, Bachiller Corral J y col. Large granular lymphocyte leukemia as a complication of rheumatoid arthritis. Reumatol Clín (Engl Ed). 2012;8(6):365–7. Disponible en: http://dx.doi.org/10.1016/j.reumae.2011.12.004.