Fiebre de origen desconocido como presentación de linfoma intravascular de células grandes B
Fever of unknown origin as a presentation of intravascular large B-cell lymphoma
Altube A1, Sanzo M,2 Nocetti E3, Grinspon S4, Mazzocchi O4, Contardo D4, Carbia C3, Brodsky A3, Díaz de la Fuente F2, Sciaccaluga D2, Cabral C2, Malusardi C1, Auat M1, Cordini G3, Martínez E3.
1 Laboratorio de Citometría de Flujo, División Hematología, Hospital de Clínicas José de San Martín, Universidad de Buenos Aires.
2 Departamento de Patología, Hospital de Clínicas José de San Martín, Universidad de Buenos Aires.
3 División Hematología, Hospital de Clínicas José de San Martín, Universidad de Buenos Aires.
4 Departamento de Medicina Interna, Hospital de Clínicas José de San Martín, Universidad de Buenos Aires.
|
Altube Alejandra |
0000-0003-4380-6779 |
|
Sanzo Micaela |
0009-0007-3543-2929 |
|
Nocetti Emilia |
0009-0005-9553-480X |
|
Grinspon Sebastián |
0009-0005-0485-0867 |
|
Mazzocchi Octavio |
0000-0002-3623-5675 |
|
Contardo Damian |
0000-0001-9509-0990 |
|
Brodsky Andres |
0009-0004-5333-5792 |
|
Diaz de la Fuente Florencia |
0009-0006-0625-5110 |
|
Sciaccaluga Dolores |
0009-0008-5031-2383 |
|
Cabral Cecilia |
0000-0003-3311-0401 |
|
Malusardi Cecilia |
0000-0002-7287-8539 |
|
Auat Mariangeles |
0000-0001-9312-9249 |
|
Cordini Gregorio |
0000-0001-9107-5996 |
|
Martínez Enriqueta |
0009-0001-7704-3362 |
Autor responsable: altubea@hospitaldeclinicas.uba.ar
Palabras claves: fiebre de origen desconocido,
linfoma intravascular,
anemia.
Keywords: fever of unknown origin,
intravascular lymphoma,
anemia.
Resumen
El linfoma intravascular de células grandes B (IVLBCL por sus siglas en inglés) es un linfoma de difícil diagnóstico debido a la ausencia de signos y síntomas patognomónicos, la ausencia de adenopatías y la presencia de manifestaciones clínicas inespecíficas que incluyen fiebre de origen desconocido, malestar general y anemia. Conocer las diferentes formas de presentación permitirá un diagnóstico precoz y, en consecuencia, un mejor pronóstico para estos pacientes. Este estudio retrospectivo analizó los síntomas de presentación, las características clínicas y de laboratorio, el comportamiento y el manejo terapéutico de 4 pacientes diagnosticados con IVLBCL en nuestro hospital durante los últimos 15 años. Todos los pacientes presentaron fiebre de origen desconocido, ausencia de linfadenopatías, anemia, aumento de ferritina sérica, marcado aumento de LDH y de β2-microglobulina, y 3 de 4 pacientes presentaron monocitosis. En 3 de 4 casos se observaron síntomas neurológicos. Si bien la tomografía por emisión de positrones mostró captación en médula ósea en 2 de los 3 pacientes donde se realizó este estudio, sólo 1 de estos pacientes presentó compromiso por histología. En 2 pacientes el diagnóstico de IVLBCL se realizó por biopsia aleatoria de piel de apariencia normal (RSB: random skin biopsy). La presencia de fiebre de origen desconocido justifica la toma de RSB, ya que ha permitido realizar diagnósticos tempranos de este tipo de linfoma. Conocer las distintas formas de presentación de esta enfermedad resultará en un diagnóstico precoz y, en consecuencia, en mejores resultados para estos pacientes.
Abstract
Intravascular large B-cell lymphoma (IVLBCL) presents diagnostic challenges due to the absence of pathognomonic signs and symptoms, lack of lymphadenopathy, and the presence of nonspecific clinical manifestations such as fever of unknown origin, malaise, and anemia. Recognizing its diverse clinical presentations is essential for early diagnosis and improved patient outcomes.
This retrospective study analyzed the presenting symptoms, clinical and laboratory findings, clinical course and treatment strategies in four patients diagnosed with IVLBCL at our institution over the past 15 years. All patients presented with fever of unknown origin, absence of lymphadenopathy, anemia, elevated serum ferritin, markedly increased levels of lactate dehydrogenase and β2-microglobulin, and monocytosis in three of the four cases. Neurological symptoms were also observed in three patients.
Positron emission tomography (PET) showed bone marrow uptake in two of the three patients who underwent the scan; however, histologic confirmation of marrow involvement was found in only one case. In two patients, the diagnosis was established via random skin biopsy of clinically normal-appearing skin. Given the presence of fever of unknown origin, random skin biopsy should be considered, as it may facilitate early identification of IVLBCL.
Increased awareness of the varied clinical presentations of IVLBCL can lead to earlier diagnosis and, consequently, better outcomes for these patients.
Introducción
El linfoma intravascular de células grandes B (IVLBCL por sus siglas en inglés) es un linfoma extranodal, raro y agresivo. Se caracteriza por el crecimiento de linfocitos clonales dentro de la luz de vasos sanguíneos de pequeño y mediano tamaño. La Organización Mundial de la Salud (OMS) lo clasifica como un subtipo de linfoma de células grandes B y reconoce tres variantes: la variante clásica, la cutánea y la asociada a síndrome hemofagocítico. El diagnóstico de IVLBCL es un desafío clínico debido a que suele presentarse con una sintomatología inespecífica que incluye fiebre de origen desconocido, malestar general y anemia. La variabilidad de los síntomas es debida a la oclusión de los pequeños vasos en los distintos órganos por las células neoplásicas(1). La ausencia de adenopatías y la rara afectación de sangre periférica hacen aún más difícil su diagnóstico, y un retraso en éste, a menudo, resulta en desenlaces fatales(2). Este linfoma es considerado una enfermedad sistémica, y conocer sus distintas formas de presentación resultará en un diagnóstico precoz y, en consecuencia, en mejores resultados para estos pacientes.
Esta publicación describe los síntomas de presentación, las características clínicas y de laboratorio, así como la evolución y el manejo terapéutico de 4 pacientes con IVLBCL diagnosticados en nuestro hospital durante los últimos 15 años.
Materiales y métodos
Pacientes
En esta serie de casos, se realizó una revisión retrospectiva de los registros de pacientes diagnosticados en nuestro hospital con IVLBCL según los criterios de la OMS, entre junio de 2010 y enero de 2025. Para cada paciente se recopilaron datos sobre edad, sexo, características clínicas y de laboratorio, tratamiento recibido y respuesta obtenida.
Resultados
Un total de 4 pacientes fueron diagnosticados con IVLBCL. No hubo prevalencia de sexo y la edad promedio fue de 59,5 años, con una mediana de 61,5 años (rango, 49-66 años). El diagnóstico histopatológico se realizó con biopsia de piel en dos pacientes y biopsia de pulmón y riñón en los otros dos (Figura 1). Las características clínicas basales de los pacientes al momento del diagnóstico se presentan en la tabla 1.
Todos los pacientes presentaron fiebre de origen desconocido. No se detectaron linfadenopatías, esplenomegalia ni hepatomegalia en ninguno de ellos.
Las alteraciones hematológicas incluyeron anemia en todos los casos y trombocitopenia en un solo caso. Ninguno de los pacientes presentó linfocitosis, sin embargo, 3 de los 4 presentaban monocitosis. Todos los casos presentaron valores elevados de LDH, ferritina sérica y PCR. Los valores de VSG resultaron elevados en 3 de 4 pacientes, en uno de los casos no pudo hallarse el dato. Se detectó hipoalbuminemia en 3 de 4 pacientes. El valor de creatinina se encontró levemente aumentado sólo en uno de los casos y corresponde al paciente con sitio de diagnóstico inicial en pulmón, pero cabe señalar que este paciente presentó recaída con compromiso renal. En el paciente con diagnóstico inicial a través de biopsia renal el valor de creatinina fue normal y la biopsia renal se realizó por haber presentado una lesión hipercaptante por PET-TC (tomografía por emisión de positrones con 18F-fluorodesoxiglucosa - tomografía computada) a dicho nivel. Al momento del diagnóstico, la FAL se halló aumentada en la mitad de los casos y la GGT en 2 de los 3 casos en que fue estudiada. Los niveles de β2-microglobulina estuvieron elevados en todos los pacientes.
El estudio de medula ósea (MO) evidenció compromiso por linfoma sólo en uno de los casos, tanto por citometría de flujo multiparamétrica (CFM) como por estudio histopatológico, con un porcentaje de infiltración por linfocitos clonales del 3% sobre la celularidad total. Se observaron imágenes de hemofagocitosis en la mitad de los pacientes.
Al momento del diagnóstico sólo en un caso se detectaron linfocitos clonales en sangre periférica (SP) por CFM. En otro de los pacientes la detección fue al momento de la recaída.
Se realizó biopsia aleatoria de piel de apariencia normal (RSB: random skin biopsy) en 3 de 4 pacientes y el diagnóstico de IVLBCL se hizo en 2 de estas 3 muestras. Al examen físico, uno de estos pacientes no evidenció compromiso cutáneo; por el contrario, el otro paciente presentó “piel de naranja” en la región abdominal infraumbilical, incluso, este paciente presentó descamación de la piel post tratamiento.
Los síntomas neurológicos estuvieron presentes en 3 de 4 pacientes y las manifestaciones clínicas fueron: afasia de expresión, apraxia y bradipsiquia; mononeuropatía múltiple constatada por electromiografía; neuropatía sensitiva de miembros inferiores y alteración transitoria en la percepción de los colores. El estudio de líquido cefalorraquídeo (LCR) por CFM se realizó en 3 pacientes. De éstos, dos habían presentado síntomas neurológicos y en ninguno de los tres se detectaron linfocitos clonales. La resonancia magnética nuclear (RMN) de cerebro mostró múltiples imágenes de isquemia en el paciente con apraxia y bradipsiquia, pero sólo cuando estos síntomas progresaron. En el paciente con neuropatía sensitiva y percepción cromática alterada la RMN sólo se evidenciaron lesiones isquémicas que no pudieron explicar estos cambios en la visión. Debido a la inespecificidad de este síntoma y a que el evento fue transitorio, estas imágenes se interpretaron como cambios isquémicos propios de la edad, por lo que sólo recibió quimioterapia profiláctica para sistema nervioso central (SNC).
Los síntomas respiratorios estuvieron presentes en la mitad de los pacientes y las manifestaciones clínicas fueron, en uno de los casos, tos no productiva sin hallazgos patológicos en el PET-TC y en el otro caso disnea progresiva con disminución de la saturación de oxígeno y prueba de difusión de monóxido de carbono (DLCO) disminuida en grado severo. Si bien la TC de tórax de éste último evidenció aumento de la atenuación en vidrio esmerilado a nivel de parénquima pulmonar bilateral, la biopsia de pulmón que condujo al diagnóstico se realizó debido a la alteración de las pruebas funcionales respiratorias y no al hallazgo tomográfico, que en un primer momento había sido interpretado como secundario a un proceso infeccioso.
Con respecto a los hallazgos en el PET-TC, el paciente 1 mostró imágenes de concentración patológica del radiotrazador en riñón, hígado, MO, bazo y lóbulo tiroideo izquierdo. En la paciente 2 en endometrio, vagina y ambas amígdalas, y fueron interpretadas como inespecíficas/reactivas. En el paciente 3 no se realizó PET al momento del diagnóstico. Por último, en la paciente 4 se evidenció incremento de la actividad metabólica de la MO en esqueleto axial y apendicular. En la tabla 1 se presenta la comparación del compromiso de MO por PET-TC versus el estudio histopatológico.
Todos los pacientes fueron tratados con esquema R-CHOP (rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona) y terapia intratecal con metotrexato (MTX), citarabina y dexametasona, a excepción de un paciente que, además, recibió profilaxis con MTX sistémico. Cabe aclarar que el paciente con compromiso de SNC se encontraba en plan de recibir tratamiento con alcance a parénquima cerebral, pero falleció luego del primer ciclo de R-CHOP. Al momento de escribir este manuscrito sólo uno de los pacientes continuaba libre de enfermedad (el tiempo de seguimiento actual es de 33 meses), el resto de los pacientes fallecieron por causas relacionadas al linfoma, a saber, falla en la etapa de inducción y causas relacionadas al trasplante autólogo de células madre. La sobrevida libre de progresión del paciente 3 fue de doce meses hasta la recaída, luego de la cual realizó quimioterapia de rescate seguido de consolidación con trasplante autólogo de médula ósea falleciendo a los dos meses posteriores al trasplante.
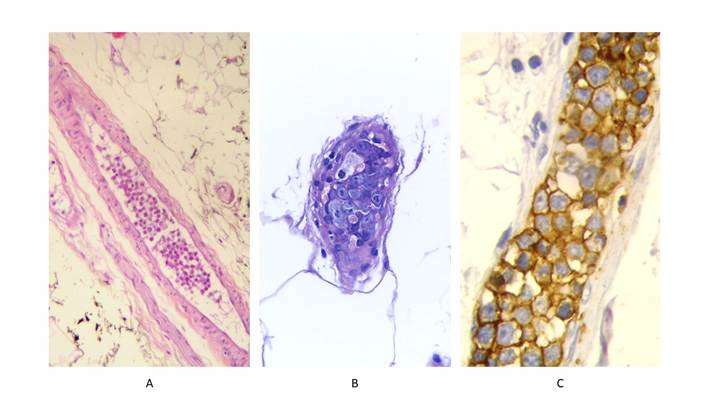
Figura 1. Análisis anatomopatológico. En A tejido adiposo perirrenal y en B tejido celular subcutáneo. Células linfoides neoplásicas de mediano y gran tamaño en vasos de pequeño y mediano calibre (A: H&E, 200X y B: H&E, 400X). Los linfocitos neoplásicos presentan irregularidad nuclear y nucléolos evidentes (B: H&E, 400X), también evidencian positividad de manera intensa con CD20 (C: inmunohistoquímica, 400X). H&E: hematoxilina-eosina.
Discusión
Este estudio presenta la experiencia de un centro de salud en el diagnóstico y tratamiento de pacientes con IVLBCL. El mal pronóstico de esta enfermedad se debe en parte a la demora en el diagnóstico relacionada con su presentación clínica variable e inespecífica(3).
La mediana de edad de presentación de nuestros pacientes fue, en promedio, 9 años menor a la reportada en las series occidental(4) y asiática(2). No hubo diferencia en cuanto a la prevalencia de género, al igual que lo descripto en estas series. Todos nuestros pacientes presentaron la variante clásica del IVLBCL.
La fiebre de origen desconocido, el síntoma sistémico más frecuentemente reportado, estuvo presente en todos nuestros pacientes, así como los valores elevados de LDH y la ausencia de linfadenopatías. La fiebre de origen desconocido, per se, es una indicación para realizar biopsia de MO, sin embargo, debe destacarse que sólo en uno de nuestros pacientes se encontró compromiso en el estudio histopatológico. Estos resultados se aproximan más a lo reportado por Ferreri et al.(4) en la serie occidental, donde el compromiso de MO fue del 30%, que a lo reportado recientemente por Matsue et al.(2) en la serie oriental, donde el compromiso fue del 60%.
El diagnóstico de IVLBCL mediante RSB ha permitido realizar diagnósticos tempranos de este linfoma con alta sensibilidad y especificidad(2,5). Las manifestaciones cutáneas son heterogéneas, observándose lesiones que van desde “piel de naranja” hasta tumores o nódulos ulcerados. Al momento del diagnóstico el compromiso cutáneo sólo está presente en el 40% de los casos; por esta razón, y en base a nuestros resultados y a las recomendaciones de expertos, la presencia de fiebre de origen desconocido justifica la toma de RSB(6), asegurándose que la muestra obtenida incluya tejido celular subcutáneo.
Al igual que lo reportado en la literatura, los parámetros de laboratorio más frecuentemente alterados en nuestros pacientes fueron anemia, aumento de β2-microglobulina, hipoalbuminemia y marcado aumento de LDH, ferritina sérica, VSG y PCR. Es importante destacar que la monocitosis es la primera vez que se reporta en una serie de casos de pacientes con IVLBCL.
Luego de los síntomas B, los síntomas neurológicos son los más frecuentemente reportados en los pacientes con IVLBCL(7). Si bien la RMN puede ser útil para evaluar el compromiso neurológico, no existen signos patognomónicos, y muchos de los hallazgos reportados son inespecíficos y están relacionados a cambios isquémicos, que probablemente estarían producidos por la oclusión de los pequeños capilares cerebrales por parte de las células neoplásicas(2, 6).
El compromiso renal en el IVBCL se reporta en el 2-13% de los casos, y la presentación clínica puede ir desde proteinuria leve a síndrome nefrótico, insuficiencia renal, aumento del tamaño de los riñones o lesión renal(8). En nuestra serie, sólo un paciente presentó lesión renal al momento del diagnóstico con función renal conservada y otro paciente presentó proteinuria en rango no nefrótico, pero al momento de la recaída.
Greer et al.(7) reportan que los síntomas respiratorios están presentes en el 21% de los pacientes diagnosticados con IVLBCL, y pueden incluir disnea, tos o insuficiencia respiratoria con estudios por imágenes normales. En ausencia de enfermedad parenquimatosa pulmonar, la medición disminuida de la DLCO sugiere enfermedad vascular pulmonar y, si además se acompaña de síntomas sistémicos, debería considerarse este tipo de linfoma que afecta a la microvasculatura(9).
Si bien el PET-TC mostró captación patológica en médula ósea en 2 de 3 pacientes, sólo en uno se confirmó compromiso por histopatología. Se requerirán estudios más específicos para comprender el contexto reactivo en el que se desarrolla este linfoma.
Ante un paciente con fiebre de origen desconocido, es importante considerar al IVLBCL dentro de los diagnósticos diferenciales y conocer la variabilidad de manifestaciones clínicas que pueden acompañar a este linfoma. Hacer un diagnóstico precoz impacta directamente en el pronóstico del paciente.
Tabla 1. Características clínicas basales de los pacientes al momento del diagnóstico
|
Paciente 1 |
Paciente 2 |
Paciente 3 |
Paciente 4 |
||
|
Edad |
57 |
66 |
49 |
66 |
|
|
Sexo |
Hombre |
Mujer |
Hombre |
Mujer |
|
|
Sitio de diagnóstico |
Riñón |
Piel |
Pulmón |
Piel |
|
|
Hallazgos de laboratorio |
|||||
|
Recuento GB (x10⁹/L) |
11.00 |
6.55 |
7.69 |
9.66 |
|
|
Neutrófilos (%) |
62 |
42 |
56 |
65 |
|
|
Linfocitos (%) |
32 |
40 |
17 |
13 |
|
|
Monocitos (%) |
5 |
15 |
23 |
16 |
|
|
Hemoglobina (g/L) |
111 |
111 |
105 |
85 |
|
|
Recuento plaquetas (x10⁹/L) |
44 |
329 |
227 |
244 |
|
|
LDH (UI/L) |
855 |
878 |
1788 |
702 |
|
|
PCR (mg/L) |
28 |
8 |
29 |
186 |
|
|
VSG (mm/h) |
extraviado |
76 |
79 |
136 |
|
|
Ferritina (μg/L) |
˃1500 |
˃1500 |
1260 |
˃1600 |
|
|
Albúmina (g/L) |
34 |
31 |
37 |
27 |
|
|
Bilirrubina total (mg/L) |
8 |
5 |
5 |
5 |
|
|
Creatinina (mg/L) |
5.9 |
7.5 |
10 |
8.5 |
|
|
FAL (IU/L) |
316 |
83 |
77 |
123 |
|
|
GGT (IU/L) |
40 |
no testeado |
33 |
85 |
|
|
β2-microglobulina (mg/L) |
3.1 |
5.8 |
6.1 |
4.3 |
|
|
Síntomas B |
|||||
|
Fiebre |
Sí |
Sí |
Sí |
Sí |
|
|
Sudoración nocturna |
Sí |
No |
Sí |
No |
|
|
Pérdida de peso |
Sí |
No |
Sí |
No |
|
|
Linfoadenopatías |
No |
No |
No |
No |
|
|
Hepatomegalia |
No |
No |
No |
No |
|
|
Esplenomegalia |
No |
No |
No |
No |
|
|
Signos y síntomas respiratorios |
Tos no productiva |
No |
Disnea |
No |
|
|
Signos y síntomas neurológicos |
Parestesia y debilidad de miembros inferiores. Mononeuropatía múltiple |
Afasia de expresión, apraxia, bradipsiquia |
No |
Neuropatía sensitiva. Cambios visuales |
|
|
Lesiones en piel |
No detectables |
No detectables |
No detectables |
Sí |
|
|
Hemofagocitosis en MO |
Sí |
No |
No |
Sí |
|
|
Compromiso de MO (%) |
Sí (3% de infiltración) |
No detectable |
No detectable |
No detectable |
|
|
Células tumorales en SP por CFM |
No detectable |
No detectable |
No detectable |
0,27% |
|
|
PET-TC (captación) |
Riñón, MO, hígado, bazo y glándula tiroides |
Endometrio, vagina y amígdalas |
no testeado |
MO |
|
|
Quimioterapia |
RCHOP + TIT |
RCHOP + TIT |
RCHOP + TIT |
RCHOP + TIT+MTX prof. |
|
|
SLP |
1 mes |
1 mes |
12 meses |
libre de enfermedad |
|
Tabla 1. En azul parámetros fuera de los valores de referencia y hallazgos anormales. Valores de referencia: recuento GB (glóbulos blancos): 4-11 x10⁹/L, neutrófilos: 43-65 %, linfocitos: 21-46 %, monocitos: 5-12 %, hemoglobina: 120-140 g/L. Recuento plaquetas: 150-450 x10⁹/L, LDH (lactato deshidrogenasa): 135-250 IU/L, PCR (proteína C reactiva): menor a 5 mg/L, VSG (velocidad de sedimentación globular): hasta 15 mm/h, ferritina sérica: 10-120 μg/L, albúmina: 35-52 g/L, bilirrubina total: hasta 10 mg/L, creatinina: 5.0-9.0 mg/L, FAL (fosfatasa alcalina): 35-104 IU/L, GGT (gamma glutamil transpeptidasa): 5-36 IU/L, β2-microglobulina: 0.8-2.2 mg/L.
TIT: triple terapia intratecal. MTX prof.: metotrexato profilaxis. SLP: sobrevida libre de progresión.
Bibliografía
1. Murase T, Yamaguchi M, Suzuki R et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
2. Matsue K, Abe Y, Narita K et al. Diagnosis of intravascular large B cell lymphoma: novel insights into clinicopathological features from 42 patients at a single institution over 20 years. Br J Haematol. 2019;187(3):328-336.
3. Nakamura S, Ponzoni M, Campo E. lntravascular large B-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL et al., (Eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition). International Agency for Research on Cancer (IARC); 2017: 319-320.
4. Ferreri AJ, Dognini GP, Campo E et al. Variations in clinical presentation, frequency of hemophagocytosis and clinical behavior of intravascular lymphoma diagnosed in different geographical regions. Haematologica. 2007;92(4):486-492.
5. Matsue K, Abe Y, Kitadate A et al. Sensitivity and specificity of incisional random skin biopsy for diagnosis of intravascular large B-cell lymphoma. Blood. 2019;133(11):1257-1259.
6. Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks. Blood. 2018;132(15):1561-1567.
7. Geer M, Roberts E, Shango M, Till BG, Smith SD, Abbas H, Hill BT, Kaplan J, Barr PM, Caimi P, Stephens DM, Lin E, Herrera AF, Rosenbaum E, Amengual JE, Boonstra PS, Devata S, Wilcox RA, Kaminski MS, Phillips TJ. Multicenter retrospective study of intravascular large B-cell lymphoma treated at academic institutions within the United States. British Journal of Haematology. 2019; 186(2): 255-262.
8. Pothen L, Aydin S, Camboni A, Hainaut P. Nephrotic syndrome without kidney injury revealing intravascular large B cell lymphoma. BMJ Case Rep. 2019;12(6):e229359.
9. Nguyen TT, Sekiguchi H, Yi ES, Ryu JH. Occult Diffuse Neoplasm in the Lungs: Intravascular Large B-Cell Lymphoma. American Journal of Medicine. 2021; 134(7): 926-9.