Estudios complementarios: hemograma normal, fórmula leucocitaria conservada, LDH y hepatogramas normales. Serologías virales (hepatitis C, hepatitis B, HIV, HTLV) negativas, Chagas y VDRL negativas. Perfil tiroideo, vitamina B12 y ácido fólico normales. Estudios de autoinmunidad: C3/C4 normales, FAN negativo, proteinograma electroforético sin pico monoclonal, IgG, IgM normales, perfil celíaco negativo. PPD negativa. LCR: leucocitos a predominio mononuclear con hiperproteinorraquia y ácido láctico elevado. Cultivos (Koch, gérmenes comunes, micológico, virológico) negativos.
Estudios de imagen. Tomografía cuello-tórax-abdomen-pelvis: esplenomegalia leve. Resonancia magnética de cerebro y columna completa con contraste EV: refuerzo y engrosamiento de las raíces de la cola de caballo, asociado a refuerzo ependimario y de pares craneales con áreas pseudonodulares. Ecografía testicular y fondo de ojo: normales. Ecocardiograma transtorácico, sin hallazgos relevantes.
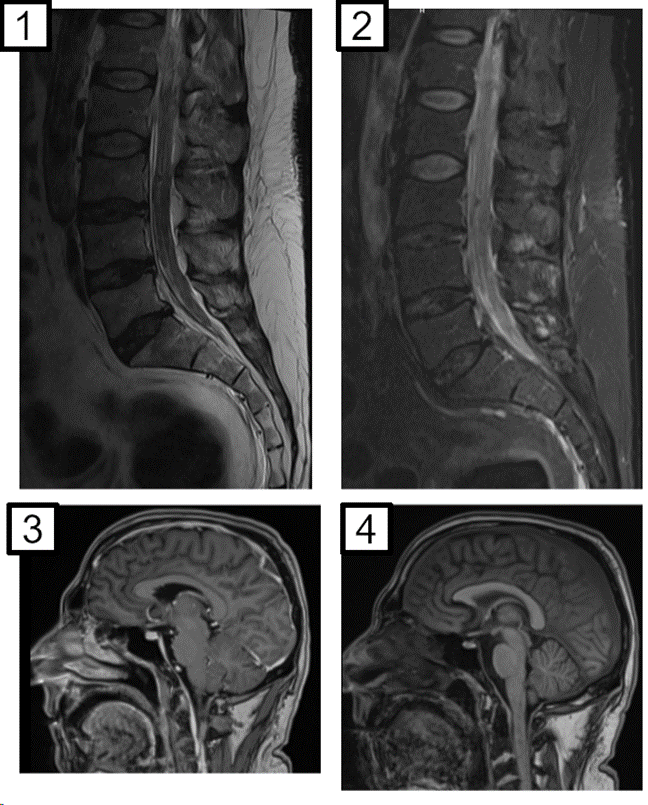
Figura 1. Resonancia magnética de columna lumbosacra en un corte sagital en secuencia T2 (1) y T1 con contraste EV (2), donde se observa refuerzo y engrosamiento de las raíces de la cola de caballo. En los cortes sagitales de encéfalo en secuencias T1 con contraste (3) y sin (4) se observa realce paquimeníngeo a predominio frontal.
Ante esta sospecha diagnóstica, el paciente recibe tratamiento con altas dosis de metilprednisolona, sin presentar mejoría franca. Entre los múltiples diagnósticos diferenciales se plantea la posibilidad de tratarse de un síndrome linfoproliferativo con afectación meníngea. Por esto, se realiza citometría de flujo de LCR por punción transcisternal, la cual informa: presencia de células de gran tamaño y complejidad, CD45++, CD19++, CD20++, CD5(-), CD38+, CD10-/+, CD22+, restricción de las Igs de superficie, cadena liviana, kappa, vinculables a linfocitos B clonales, hallazgos compatibles con compromiso del SNC por linfoma no Hodgkin fenotipo B con componente de células de gran tamaño.
Se complementa estudio con biopsia de meninges, en la cual se halla sólo tejido fibroconjuntivo denso, sin alteraciones. Pese a este resultado, se decide postergar nueva toma de biopsia en el contexto del cuadro clínico del paciente, priorizando el inicio del tratamiento onco-específico.
Sujetos a la disponibilidad de drogas y recursos en aquel momento, se inicia esquema rituximab-metotrexato-citarabina, con un primer ciclo bien tolerado y sin complicaciones intra-administración. En los días subsiguientes el paciente refiere mejoría parcial de síntomas, adquiriendo la capacidad de sentarse por sí mismo y mejorando cuadro de paresia de miembros inferiores. Pese a presentar citopenias marcadas, sólo requiere soporte con factores estimulantes de colonias. Una semana luego de finalizado el ciclo y con parámetros de laboratorios aptos para manejo ambulatorio, se otorga su alta con control pautado en los días subsiguientes.
Reingresa a las 24 horas por cuadro de neutropenia febril a foco abdominal, el cual a los pocos días progresa a shock séptico, con requerimiento de manejo en unidad cerrada con necesidad de IOT/AVM, soporte transfusional de glóbulos rojos y plaquetas, factores estimulantes de colonias, tratamiento antibiótico de amplio espectro y vasopresores. Finalmente, el paciente fallece por falla multiorgánica secundaria a shock séptico refractario.
Discusión
Uno de los diagnósticos principales sobre los que se trabajó al principio fue la CIDP. Las características típicas de la CIDP incluyen ausencia de una enfermedad asociada, historia frecuente de infecciones y tendencia al compromiso craneal, troncal, proximal y distal de los miembros, con velocidades de conducción disminuidas en los nervios periféricos, sobre todo a nivel proximal. Las características patológicas incluyen edema de serosas, infiltrados de células mononucleares (sobre todo en áreas perivasculares, pero sin evidencia de vasculitis), desmielinización segmentaria inducida por macrófagos y neuritis hipertrófica(3). Según la última revisión del consenso sobre CIDP(5), actualmente se clasifican en:
- CIDP típica: debilidad progresiva o reincidente, simétrica, proximal y distal de miembros superiores e inferiores, con compromiso sensitivo de al menos dos miembros, con un tiempo de evolución mayor a 8 semanas y reflejos ausentes o reducidos en todos los miembros.
- CIDP variantes: se subdividen a su vez en:
- CIDP distal: debilidad muscular y pérdida sensitiva en miembros inferiores.
- CIDP multifocal: debilidad muscular y pérdida sensitiva en un patrón multifocal, usualmente asimétrico, con predominio en miembros superiores, con compromiso de más de un miembro.
- CIDP focal: debilidad muscular y perdida sensitiva en sólo un miembro
- CIDP motora: signos y síntomas motores, pero sin compromiso sensitivo.
- CIDP sensitiva: signos y síntomas sensitivos, pero sin compromiso motor.
En el caso de este paciente, se concluyó que podría corresponder a la variante distal por la presentación en miembros inferiores, sin afectar los superiores. Los objetivos del diagnóstico y manejo son controlar la inflamación y prevenir pérdidas axonales que pueden llevar a la disfuncionalidad. Aunque en algunos casos el curso es monofásico con recuperación completa, en otros puede ser más lento y progresivo, o con un perfil de recaídas-remisiones, resultando en una morbilidad prolongada y, en ocasiones, en una discapacidad permanente(4).
Posteriormente, mediante la citometría de flujo de LCR se arriba al diagnóstico de linfoma primario de sistema nervioso central (LPSNC). Es una enfermedad maligna agresiva exclusiva del sistema nervioso central (SNC), o sea el parénquima cerebral, médula espinal, pares craneales y/o meninges. Este linfoma representa el 4% de las neoplasias intracraneales y 4-6% de todos los linfomas extranodales, y hay registros de la incidencia en aumento en pacientes inmunocompetentes. El LPSNC presenta características clínicas y biológicas que implican un desafío diagnóstico y terapéutico para clínicos y científicos multidisciplinarios(6).
Su evolución sigue siendo insatisfactoria comparada con pacientes con linfomas extra-SNC de un subtipo y estadio similar, y varios factores previenen el avance terapéutico. Primero, el conocimiento molecular y biológico es insignificante comparado con otros linfomas, lo que limita la identificación de dianas terapéuticas. Segundo, los pacientes con LPSNC usualmente presentan peores condiciones clínicas y estado funcional que otros pacientes con otros linfomas(6).
El tratamiento moderno para pacientes con LPSNC incluye una etapa de inducción y una de consolidación. La primera consiste en poliquimioterapia con drogas de alta biodisponibilidad en SNC como corticoides y agentes alquilantes, y drogas con baja capacidad de penetrancia de la barrera hemato-encefálica pero administradas en altas dosis, como el metotrexato y citarabina. La poliquimioterapia basada en altas dosis de metotrexato seguida de una consolidación con radioterapia de todo el cerebro han sido la estrategia más comúnmente adoptada(7). El rituximab, anticuerpo monoclonal anti-CD20, se asoció con una respuesta del 42% en una pequeña serie de pacientes con LPSNC recaído o refractario(8). En este caso, se adoptó la estrategia de rituximab-metotrexato-citarabina.
Se observó que el paciente a las 48 horas de haber finalizado el metotrexato presentó los primeros signos de mejoría al recuperar fuerza y sensibilidad en los miembros inferiores, bajo el apoyo de la asistencia diaria por kinesiología motora. Al momento del alta había logrado sentarse por su cuenta, no pudiendo aún mantenerse en pie. Desafortunadamente no pudo continuar su seguimiento y tratamiento con los subsiguientes ciclos debido al desafortunado desenlace de sus complicaciones infectológicas posteriores.
Conclusiones
Se presenta el caso de un masculino de 48 años cuya manifestación inicial del linfoma primario del sistema nervioso central fue una polirradiculopatía crónica inflamatoria, una forma muy rara y poco prevalente. Fue tratado inicialmente con altas dosis de corticoides, sin respuesta. Posterior a obtener el diagnóstico, se benefició sintomáticamente de haber recibido poliquimioterapia dirigida. Sin embargo, no pudo evaluarse la respuesta y seguimiento a largo plazo debido al desenlace brusco del caso, consecuencia de las complicaciones secundarias de la inmunosupresión del tratamiento usado.
Bibliografía
1. Vallat JM, Sommer C, Magy L. Chronic inflammatory demyelinating polyradiculoneuropathy: diagnostic and therapeutic challenges for a treatable condition. Lancet Neurol. 2010 Apr;9(4):402-12. doi: 10.1016/S1474-4422(10)70041-7.
2. Hochberg FH, Miller DC. Primary central nervous system lymphoma. J Neurosurg. 1988 Jun;68(6):835-53. doi: 10.3171/jns.1988.68.6.0835.
3. Dyck PJ, Lais AC, Ohta M, Bastron JA, Okazaki H, Groover RV. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc. 1975 Nov;50(11):621-37.
4. Silwal A, Pitt M, Phadke R, Mankad K, Davison JE, Rossor A, DeVile C, Reilly MM, Manzur AY, Muntoni F, Munot P. Clinical spectrum, treatment and outcome of children with suspected diagnosis of chronic inflammatory demyelinating polyradiculoneuropathy. Neuromuscul Disord. 2018 Sep;28(9):757-765. doi: 10.1016/j.nmd.2018.06.001.
5. Van den Bergh PYK, van Doorn PA, Hadden RDM et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force—Second revision. J Peripher Nerv Syst. 2021; 26(3): 242–268. https://doi.org/10.1111/jns.12455
6. Ferreri AJ. How I treat primary CNS lymphoma. Blood. 2011 Jul 21;118(3):510-22. doi: 10.1182/blood-2011-03-321349.
7. Ferreri AJ, Cwynarski K, Pulczynski E et al; International Extranodal Lymphoma Study Group (IELSG). Chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) in patients with primary CNS lymphoma: results of the first randomization of the International Extranodal Lymphoma Study Group-32 (IELSG32) phase 2 trial. Lancet Haematol. 2016 May;3(5):e217-27. doi: 10.1016/S2352-3026(16)00036-3.
8. Batchelor TT, Grossman SA, Mikkelsen T, Ye X, Desideri S, Lesser GJ. Rituximab monotherapy for patients with recurrent primary CNS lymphoma. Neurology. 2011 Mar 8;76(10):929-30. doi: 10.1212/WNL.0b013e31820f2d94.