Accidente cerebrovascular isquémico secundario a deficiencia de antitrombina III: reporte de caso clínico
Ischemic stroke secondary to antithrombin III deficiency: a case report
Wilches Meza A1; Navarro J1; García Osorio A1,3; Rodríguez Orozco J4; Arenas López K1; González Torres E3; Martínez Guerrero G2,3; Chaytili Gómez K3
1- Departamento de Medicina Interna. Organización Clínica Bonnadona Prevenir, Barranquilla, Atlántico. Colombia
2- Departamento de Epidemiología. Organización Clínica Bonnadona Prevenir, Barranquilla, Atlántico. Colombia
3- Semillero de Investigación. Organización Clínica Bonnadona Prevenir, Barranquilla, Atlántico. Colombia
4- Departamento de Hematología. Organización Clínica Bonnadona Prevenir, Barranquilla, Atlántico. Colombia
Alfonso Wilches Meza: https://orcid.org/0009-0000-7959-9607
José Navarro: https://orcid.org/0009-0002-5854-3238
Kellys Sofía Arenas López: https://orcid.org/0009-0009-3116-4861
José Rodríguez Orozco: https://orcid.org/0009-0005-1909-6836
Andrés García Osorio: https://orcid.org/0000-0001-5904-2369
Eliana Rosa González Torres: https://orcid.org/0009-0004-8741-582X
Gillian Martínez Guerrero: https://orcid.org/0009-0004-2087-313X
Karime Chaytili Gómez: https://orcid.org/0009-0000-7164-4311
coordepidemiologiaclinica@bonnadona.co
Palabras clave: encefalopatía,
antitrombina,
ACV.
Keywords: encephalopathy,
antithrombin,
ACV.
Resumen
Se presenta el caso de un paciente de 25 años con un accidente cerebro vascular (ACV) isquémico atípico, sin antecedentes clásicos de riesgo. Ingresó por cefalea intensa que evolucionó a paro cardiorrespiratorio. La tomografía computarizada de cerebro reportó múltiples áreas de isquemia en el cerebelo y la región occipitotemporal derecha no atribuibles a hipoperfusión post-RCP, mientras que la resonancia magnética evidenció lesiones isquémicas agudas extensas con transformación hemorrágica en lóbulos parieto-occipitales bilaterales, temporal derecho, hemisferios cerebelosos, bulbo raquídeo y otras regiones profundas. Los estudios confirmaron deficiencia grave de antitrombina III (AT) como única causa identificable. Esta proteína es clave en la regulación de la coagulación, y su déficit provoca hipercoagulabilidad. Este caso subraya la necesidad de investigar trastornos de coagulación en pacientes jóvenes con trombosis y resalta la importancia de un diagnóstico temprano y manejo multidisciplinario.
Abstract
This is the case of a 25-year-old patient with an atypical ischemic cerebrovascular accident (CVA) and no classical risk factors. The patient was admitted due to severe headache, which progressed to cardiorespiratory arrest. A brain CT scan revealed multiple ischemic areas in the cerebellum and the right occipitotemporal region, not attributable to post-CPR hypoperfusion. MRI showed extensive acute ischemic lesions with hemorrhagic transformation in the bilateral parieto-occipital lobes, right temporal lobe, cerebellar hemispheres, brainstem, and other deep regions. Studies confirmed severe antithrombin III (AT) deficiency as the only identifiable cause. This protein plays a key role in coagulation regulation, and its deficiency leads to hypercoagulability. This case underscores the importance of investigating coagulation disorders in young patients with thrombosis and highlights the need for early diagnosis and multidisciplinary management.
Introducción
El ACV es la segunda causa de muerte a nivel mundial, asociado principalmente a factores cardiovasculares, aunque también puede presentarse en pacientes con trombofilias hereditarias o adquiridas(1).
La AT es sintetizada en hígado, pulmones y riñones, y su función es inactivar de forma progresiva e irreversible a la trombina en ausencia de heparina y también a los factores de coagulación IXa, Xa, XIa y XIIa. Esta molécula está constituida por alfa-2 globulina, responsable del 50% de su actividad funcional, complementada por alfa-2 macroglobulina y alfa-2 antitripsina, que contribuyen con el otro 50%(2).
La deficiencia de AT, tiene una incidencia del 0.02 al 0.2%(3), provoca un estado de hipercoagulabilidad, aumentando el riesgo de trombosis venosa profunda, tromboflebitis y trombosis de venas cerebrales o abdominales, especialmente en pacientes con factores de riesgo como cirugías recientes, traumatismos, terapias hormonales o inmovilidad prolongada(6).
Caso clínico
Paciente masculino de 25 años, remitido bajo contexto de cefalea súbita e intensa seguido de deterioro del estado de consciencia y eventual paro cardiorrespiratorio, requiriendo ventilación mecánica invasiva con posterior traqueostomía y gastrostomía percutánea. A su ingreso se documentó sepsis de foco mixto con cultivo positivo para bacilos Gram negativos. Al examen físico, se encontraba en estado de mínima consciencia (Glasgow 7/15).
Los exámenes iniciales mostraron leucocitosis (34130/uL), anemia grado II según OMS (10.8 g/dl), tiempos de coagulación prolongados (TP 17.5 seg, INR 1.31, TPT 31.2 seg), ferritina elevada (1199 ng/ml), gases arteriales (pH 7.43, PcO2 41.5 mmHg, HCO3 27 mmol/L, BE 30 mmol/L) y alteraciones en función hepática (AST 164 U/L, ALT 355 U/L) y renal (BUN 47 mg/dl y creatinina 1.15 mg/dl). El hemocultivo reveló bacilos gram negativos sin rescate microbiológico.
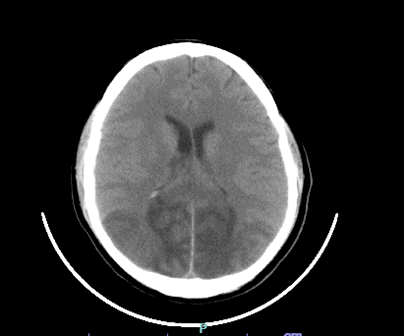
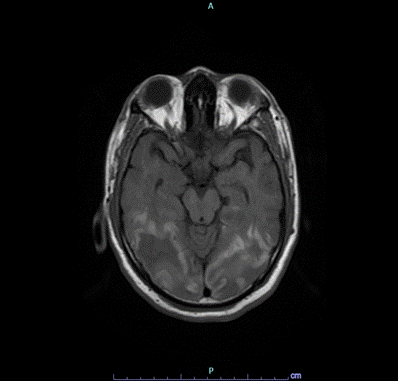
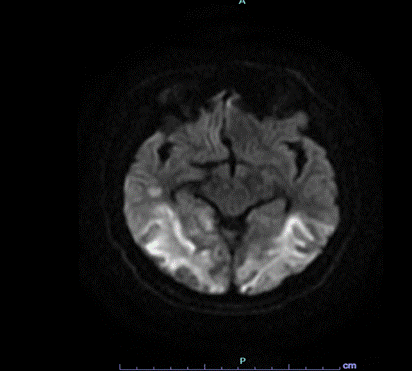
Las imágenes diagnósticas reportaron TAC de cráneo que evidenció un evento isquémico cerebeloso y occipitotemporal derecho, mientras que el ecocardiograma mostró una fracción de eyección reducida al 42%. La resonancia magnética de cerebro reportó extensas lesiones cortico-subcorticales sugestivas de patología vascular isquémica aguda, comprometiendo los lóbulos parieto-occipitales bilateralmente y temporal derecho, con signos de transformación hemorrágica, en territorio de las arterias cerebrales posteriores. Así mismo tiene lesiones de características similares en ambos hemisferios cerebelosos, bulbo raquídeo y esplenio del cuerpo calloso, como también en ganglio basal derecho. La angioresonancia no mostró alteraciones vasculares detectables.
Se inició tratamiento con antibióticos de amplio espectro y heparina de bajo peso molecular a dosis de 60 UI SC cada 12 horas. Se realizó un diagnóstico inicial en conjunto con medicina interna y neurología de infarto cerebral basilar-ACP bilateral con sospecha de trombosis basilar, disección vertebral e hipercoagulabilidad. Se realizaron múltiples estudios como anticuerpos antinucleares, anti fosfolipídicos, anticoagulante lúpico, beta 2 glicoproteína IgG e IgM en rangos normales, se estudiaron a demás otras trombofilias hereditarias incluyendo niveles de proteína C, proteína S y mutación del factor V de Leiden y mutación del gen de la protrombina.
Ante la presencia de los eventos isquémicos cerebrales en un paciente joven sin antecedentes clínicos relevantes, se inició protocolo de estudio de trombofilias adquiridas y hereditarias. Se determinaron los niveles de antitrombina III, los cuales fueron inferiores a 7.5 mg/dl (valor de referencia 12-28 mg/dl por método de inmunoturbidimetría), la medición fue repetida en 2 ocasiones con diferencia de 72 horas, confirmándose la deficiencia de déficit severo. Consideramos un factor de riesgo significativo para el desarrollo de trombosis en el contexto clínico del paciente. Tras los resultados de los estudios de laboratorio, las imágenes diagnósticas y la ausencia de otras causas en pacientes jóvenes que justificaran los hallazgos y la clínica que presentaba el paciente, se estableció como diagnóstico definitivo una encefalopatía estructural secundaria a isquemia cerebral en múltiples territorios, atribuible a una trombofilia primaria causada por un déficit severo de antitrombina III.
Tras su estabilización, el paciente fue remitido a un centro de cuidados crónicos para continuar con el manejo y la rehabilitación. Actualmente con buen desempeño, sin traqueostomía ni gastrostomía, funcionalmente independiente.
Se recomendó la evaluación genética posterior al egreso para descartar mutación en el gen SERPINC1, asociado al déficit hereditario tipo I y tipo II. El paciente se encuentra en seguimiento ambulatorio con planes de estudio para confirmar o descartar origen hereditario. Hasta el momento no disponemos de estudios genéticos.
 |
 |
|||
 |
|||
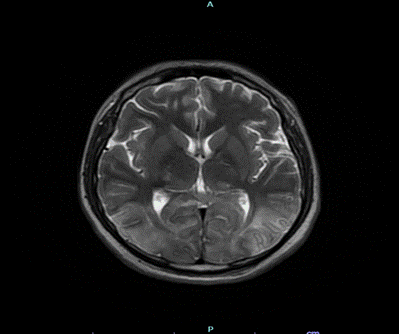
 |
 |
|||
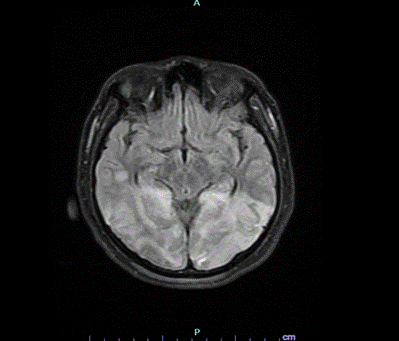 |
|||
 |
 |
||
Discusión
El déficit de antitrombina III es una condición poco frecuente pero clínicamente relevante, asociada con un alto riesgo de trombosis en diferentes contextos clínicos. Este caso subraya la importancia de considerar las trombofilias hereditarias o adquiridas en pacientes jóvenes con eventos tromboembólicos atípicos, como los descritos en este paciente crítico de 25 años(7,8).
El déficit de AT puede clasificarse en dos grandes categorías: hereditario (tipo I o tipo II) y adquirido. El déficit hereditario es poco frecuente, con una prevalencia estimada de 1 en 2,000 a 5,000 individuos, y se caracteriza por la reducción de la actividad funcional (tipo II) o de la concentración plasmática (tipo I), suficiente para predisponer a episodios trombóticos(7,8). Aunque el paciente no tenía antecedentes personales o familiares claros de trombosis, es posible que el déficit de AT hereditario se haya manifestado bajo el estrés inflamatorio severo. El déficit adquirido, por otro lado, se observa más comúnmente en condiciones como sepsis, síndrome nefrótico, insuficiencia hepática o uso prolongado de heparina no fraccionada(9,10).
La AT es un inhibidor clave de la cascada de coagulación, particularmente en la neutralización de trombina y los factores Xa y IXa. Su déficit, ya sea hereditario o adquirido, genera un estado de hipercoagulabilidad que predispone al desarrollo de trombosis en el lecho arterial y venoso. Los eventos trombóticos en estos pacientes pueden ocurrir espontáneamente o ser precipitados por factores adicionales, como infecciones severas, inmovilización o traumatismos, todos presentes en este caso(10,11).
En condiciones normales, la AT desempeña un papel crucial al inactivar de manera progresiva e irreversible a la trombina, además de otros factores de coagulación mencionados. Este proceso se lleva a cabo en conjunto con otras proteínas, como la alfa-2-globulina, que neutraliza aproximadamente el 50% de la trombina(8,10).
Aunque el déficit de AT es típicamente asociado con trombosis venosa profunda, embolismo pulmonar y trombosis de sitios inusuales (como las venas hepáticas o mesentéricas), también puede predisponer a eventos arteriales isquémicos(9,12). En este paciente, la TAC de cráneo y la resonancia evidenciaron múltiples áreas de infarto en el cerebelo y la región occipitotemporal derecha. Estas lesiones no eran compatibles con un síndrome de hipoperfusión típico del estado post-PCR, lo que planteó la sospecha de un estado protrombótico subyacente(7,11).
La isquemia cerebral en jóvenes con trombofilia puede ocurrir por embolismos paradójicos, formación de trombos in situ o por vasculopatías asociadas(9,11). En este caso, el déficit de AT probablemente contribuyó a una trombosis arterial local o a una complicación de origen embólico debido a su estado crítico. El deterioro hemodinámico y el estrés inflamatorio asociados a la sepsis pudieron amplificar el riesgo trombótico en este contexto(10,12).
La identificación del déficit en este paciente fue crítica para dirigir el manejo. Sin esta intervención, los eventos isquémicos habrían progresado, con un pronóstico considerablemente peor.
El diagnóstico del déficit de AT en este paciente subrayó la importancia de un enfoque sistemático para evaluar trombofilias en jóvenes con eventos trombóticos inexplicados(9,12). Las pruebas funcionales reportaron niveles <50% que confirmaron el déficit. Esta condición probablemente fue un factor determinante en el desarrollo de eventos isquémicos extensos, incluyendo las áreas cerebrales afectadas, lo cual motivó la intensificación del tratamiento anticoagulante con heparina de bajo peso molecular, ajustada cuidadosamente para evitar complicaciones hemorrágicas en un paciente ya críticamente enfermo(10,12).
El tratamiento con heparina de bajo peso molecular fue particularmente relevante en este caso, dada la capacidad de esta terapia para inhibir los efectos protrombóticos en el contexto de un déficit de AT. Sin embargo, en casos más severos o refractarios, se podría considerar la administración de concentrados de AT para corregir el déficit de manera más específica(7,11).
Conclusión
El déficit de antitrombina III, aunque infrecuente, debe incluirse en el diagnóstico diferencial de estados protrombóticos, particularmente en pacientes jóvenes con eventos isquémicos en contextos de sepsis o estrés crítico. Este caso enfatiza la importancia de una evaluación detallada y un enfoque multidisciplinario, crucial para un manejo efectivo y personalizado de estos casos.
Bibliografía
- García C, Martínez A, Garcia V, Ricaurte-Fajardo A, Torres I, Coral J. Actualización en diagnóstico y tratamiento del ataque cerebrovascular isquémico agudo. Univ Med. 2019;60(3). Disponible en: https://revistas.javeriana.edu.co/files-articulos/UMED/60-3%20(2019-III)/231059231008/
- Jiménez R. Antitrombina III en estados normales y patológicos. Rev Costarric Cienc Méd. 1983;4(2):39-54. Disponible en: https://www.binasss.sa.cr/revistas/rccm/v4n2/art6.pdf
- Franco-Díaz de León R; Gamboa-Solís R; Sánchez-Díaz de León JA et al. Trombosis venosa profunda de miembros inferiores asociada a una triple deficiencia enzimática: antitrombina III, proteína C y proteína S. Reporte de caso. Rev Lux Medica. 2021; 16(48). Disponible en: https://doi.org/10.33064/48lm20213335
- Tapia J. Enfermedad cerebrovascular y trombofilia. Rev Chil Neuro-psiquiatr. [Internet]. 2002 Abr [citado 2024 Nov 13] ; 40( 2 ): 37-45. Disponible en: http://dx.doi.org/10.4067/S0717-92272002000200004
- Accidente cerebrovascular isquémico. Manual MSD, versión para profesionales. [Internet]. 2023. Disponible en: https://www.msdmanuals.com/es/professional/trastornos-neurol%C3%B3gicos/accidente-cerebrovascular/accidente-cerebrovascular-isqu%C3%A9mico
- Déficit de antitrombina III. Redacción Médica. [Internet]. 2023. Disponible en: https://www.redaccionmedica.com/recursos-salud/diccionario-enfermedades/deficit-antitrombina-iii
- Patnaik MM, Moll S. Hereditary thrombophilia: Clinical syndromes and management. Mayo Clin Proc. 2008;83(5):533-544.
- Dahlbäck B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood Rev. 2008;22(3):107-116.
- Nitzki-George D, Farrow JM. Hypercoagulable states and sepsis: A review of pathophysiology and treatment. Crit Care Med. 2016;44(2):314-322.
- Di Nisio M, et al. Antithrombin deficiency and risk of thrombosis: A meta-analysis. Thromb Res. 2010;126(5):e337-e342.
- Levine MN, Lee AY, Kakkar AK. Thrombophilia in young patients: Aetiology and management. J Thromb Haemost. 2001;90(1):20-25.
- Berlot G et al. Antithrombin supplementation in sepsis: A narrative review. J Intensive Care Med. 2012;27(4):252-260.
- Zaidi A, Green L. Physiology of haemostasis. Anaesth Intensive Care Med. 2019;20(3):152-158. Disponible en: https://www.sciencedirect.com/science/article/abs/pii/S1472029919300050
- Francisco Pérez-Gómez and Ramón Bover. The New Coagulation Cascade and Its Possible Influence on the Delicate Balance Between Thrombosis and Hemorrhage. Rev Esp Cardiol. 2007;60(12):1217-9.
- Choreño-Parra JA, Carnalla-Cortés M, Guadarrama-Ortíz P. Enfermedad vascular cerebral isquémica: revisión extensa de la bibliografía para el médico de primer contacto. Med. interna Méx. [revista en la Internet]. 2019 Feb [citado 2024 Nov 16] ;35( 1 ):61-79. Disponible en: http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S0186-48662019000100061&lng=es. https://doi.org/10.24245/mim.v35i1.2212