Enfermedad de von Willebrand tipo 2M-C: un diagnóstico fugitivo. Acerca de un caso.
Type 2M-C von Willebrand disease: a fugitive diagnosis. About a case.
Woods AI1, Alberto MF1, Castera S1, Berger CS2,3, Lopez MS2,3, Martinuzzo ME2,3, Guerrero O1, Blanco AN1, Sánchez-Luceros A1,4
1Departamento de Hemostasia y Trombosis, Instituto de Investigaciones Hematológicas, Academia Nacional de Medicina de Buenos Aires, CABA, Argentina.
2Laboratorio Central, Sección Hemostasia, Hospital Italiano de Buenos Aires, CABA, Argentina.
3Departamento de Bioquímica Aplicada, Instituto Universitario del Hospital Italiano de Buenos Aires, CABA, Argentina
4Laboratorio de Hemostasia y Trombosis, Instituto de Medicina Experimental-CONICET, Academia Nacional de Medicina de Buenos Aires, CABA, Argentina
ORCID
Adriana Inés Woods PhD https://orcid.org/0000-0002-4077-1796
María Fabiana Alberto PhD https://orcid.org/0000-0001-7048-2031
Santiago Castera MS https://orcid.org/0000-0003-2751-6392
Claudio Sebastián Berger https://orcid.org/0009-0004-0460-0780
Marina Sol López MS https://orcid.org/0009-0002-4080-8250
Marta Elba Martinuzzo PhD https://orcid.org/0000-0002-1281-7144
Alicia Noemí Blanco PhD https://orcid.org/0000-0002-6933-1057
Analía Sánchez-Luceros MD, PhD https://orcid.org/0000-0002-2971-9217
Mail de contacto: adrianawoods@gmail.com
Palabras claves: biología molecular,
factor von Willebrand,
enfermedad de von Willebrand.
Keywords: von Willebrand factor,
von Willebrand disease,
diagnostic methods; molecular biology.
Resumen
La enfermedad de von Willebrand tipo 2M-C (VWD2M-C) se caracteriza por disminución de VWF:CB pero niveles normales de VWF:Ag y VWF:RCo. Este defecto es causado por variantes genéticas causantes de enfermedad (VGCE) en el dominio A3 del VWF que afectan la unión del VWF al colágeno.
Describimos los hallazgos fenotípicos y genotípicos en un hombre de 60 años con resultados de laboratorio sugestivos de VWD2, con índice de sangrado de 5, según el ISTH-BAT, sin antecedentes familiares de sangrado.
Métodos. Se realizó una evaluación hemostática que incluyó tiempo de cierre (TC) de PFA-200, FVIII, VWF:Ag, VWF:RCo, VWF:GPIbR, VWF:CB1,VWF:CB3, multímeros del VWF, estudios funcionales plaquetarios (lumi-agregometría, captación de mepacrina) y estudios genéticos de los dominios VWF-A1, A2 y A3.
Resultados. Primer estudio: el paciente presentó el PFA-200CT extremadamente prolongado con el cartucho Col/Epi y ligeramente prolongado con el cartucho Col/ADP con una relación VWF:GPIbR/VWF:Ag anormal. Segundo estudio: se observaron VWF:Ag, VWF:RCo y multímeros del VWF normales, pero niveles bajos de VWF:CB1 y VWF:CB3. Por lumi-agregometría se observaron agregación con ristocetina (RIPA, concentraciones estándar y bajas), ácido araquidónico y colágeno normales, ausencia de la segunda onda y liberación de ATP bajo estimulación con ADP y adrenalina, con contenido normal de gránulos densos plaquetarios. Por estudios genotípicos, se identificó la VGCE c.5191T>A → p.Ser1731Thr en el exón 30 (dominio A3) en heterocigosis. El paciente se diagnosticó como VWD2M-C.
Conclusión. En nuestro paciente, p.Ser1731Thr pareció ser responsable del PFA-200CT anormal. Dado que VWF:RCo y VWF:GPIbR miden la interacción VWF-plaquetas, y que VWF:CB mide la interacción VWF-colágeno, es de suma importancia realizar ambos ensayos para evitar la posible falta de diagnóstico de VWD2M-C en estos pacientes.
Summary
The type 2M-C von Willebrand disease (VWD2M-C) is characterized by a decreased VWF:CB but normal VWF and VWF:RCo levels. This defect is caused by disease-causing variants (DCVs) at the A3 domain of VWF affecting VWF-collagen binding.
We describe the phenotypic and genotypic findings in a 60-year-old male with laboratory results suggestive of VWD2 presenting an ISTH-BAT of 5, without family bleeding history.
Methods. Hemostatic assessment was performed including PFA-200 closure time (CT), FVIII, VWF:Ag, VWF:RCo, VWF:GPIbR, VWF:CB1,VWF:CB3, VWF multimers, platelet functional studies (lumi-aggregometry, mepacrine uptake), and genetic studies of VWF-A1, A2 and A3-domains.
Results. First study: the patient presented PFA-200CT extremely prolonged with Col/Epi cartridge and slightly prolonged Col/ADP one, and abnormal VWF:GPIbR/VWF:Ag ratio. Second study: the patient showed normal VWF:Ag, VWF:RCo and VWF multimers, but low VWF:CB1 and VWF:CB3 levels.
The lumi-aggregometric studies showed normal ristocetin (RIPA, standard and low concentrations), arachidonic acid and collagen induced platelet aggregation; absence of the second wave and ATP release under ADP and epinephrine stimulation, and normal content of platelets’ dense granules.
By genetic studies, the missense change c.5191T>A → p.Ser1731Thr in exon 30 (A3-domain) was identified in heterozygous state. The patient was diagnosed as VWD2M-C.
Conclusion. In our patient, p.Ser1731Thr seemed to be responsible for the abnormal PFA-200CT. Since the VWF:RCo and VWF:GPIbR measure the VWF-platelet interaction, while the VWF:CB, the VWF-collagen interaction, it is of utmost importance to perform both assays types to overcome the potential lack of VWD2M-C patients' diagnosis.
Introducción
La enfermedad de von Willebrand tipo 2M (VWD2M) es causada por variantes genéticas causales de enfermedad (VGCE) con modo de herencia autosómica y dominante, localizadas en el dominio A1 o A3 del factor von Willebrand (VWF), sin pérdida de los multímeros de alto peso molecular (HMWM) del mismo. Las VGCEs localizadas en el dominio A1 conducen a un defecto en la unión del VWF a la glicoproteína Ibα (GPIbα) plaquetaria, con disminución de los cocientes de los ensayos de actividad que miden esta interacción: VWF:RCo/VWF:Ag, VWF:GPIbR/VWF:Ag y VWF:GPIbM/VWF:Ag. Este subtipo se ha denominado VWD2M-P.
Por otro lado, las VGCE localizadas en el dominio A3 afectan la capacidad de unión del VWF al colágeno, cuyo efecto es la disminución de la unión del VWF a los colágenos tipos I y III (VWF:CB1 y VWF:CB3) subendoteliales, sin que se altere la capacidad de unión del VWF a la GPIbα plaquetaria. Por lo tanto, los niveles de VWF y VWF:RCo son normales. Este subtipo se denomina actualmente VWD2M-C.
Hay pocos trabajos descriptos que reporten este subtipo. El primer reporte del mismo(1) describe la mutación p.S1731T en el exón 30, en heterocigosis en dos pacientes (madre e hija) con epistaxis, equimosis y menorragia. Posteriormente se han publicado más hallazgos similares, asociados a la presencia de VGCEs en el dominio A3(2-6).
Nuestro objetivo es describir el caso de un varón de 60 años con diagnóstico previo de síndrome metabólico que fue remitido desde otro centro con resultados de laboratorio sugestivos de VWD2, que presentaba ISTH-BAT (International Society in Thrombosis and Haemostasis-Bleeding Assessment Tool) de 5, sin antecedentes familiares de sangrado.
Material y métodos
Los métodos utilizados en este paciente fueron:
· Estudios fenotípicos para evaluación hemostática que incluyen:
TTPA, FVIII (método de una etapa), VWF:Ag (ELISA e inmunoturbidimetría), VWF:RCo (agregometría), VWF:GPIbR (inmunoturbidimetría), VWF:CB (ELISA desarrollado) utilizando colágenos tipo 1 (VWF:CB1) y tipo 3 (VWF:CB3), perfil multimérico del VWF (electroforesis en gel de agarosa-SDS 1% y 1,7%), recuento de plaquetas en sangre entera (RP). Se calcularon los cocientes VWF:RCo/VWF:Ag, VWF:CB1/VWF:Ag, VWF:CB3/VWF:Ag y VWF:GPIbR/VWF:Ag, considerando normal un cociente >0,7, según las nuevas guías de diagnóstico publicadas recientemente(7).
· Estudios funcionales plaquetarios:
Tiempo de oclusión (TC) de PFA-200, lumi-agregometría, captación de mepacrina por citometría de flujo, RIPA a concentración estándar (1,2 mg/mL) y baja (≤0,7 mg/mL) de ristocetina.
El paciente fue estudiado en tres ocasiones para confirmar los hallazgos anormales.
· Estudios genotípicos:
El ADN genómico se extrajo de leucocitos de sangre periférica. Los exones 28 al 31 del gen VWF (dominios A1, A2 y A3) se amplificaron mediante tecnología de PCR previamente descripta(8) y se secuenciaron utilizando metodología de Sanger. Cuando se identifica una VGCE, la cadena complementaria del ADN se secuencia para confirmar su presencia.
Resultados
En un primer estudio realizado en el Hospital Italiano de Buenos Aires se detectó el PFA-200CT extremadamente prolongado con el cartucho Col/Epi y ligeramente prolongado con el cartucho Col/ADP, y una relación VWF:GPIbR/VWF:Ag anormal.
En un segundo estudio realizado en el Departamento de Hemostasia y Trombosis del Instituto de Investigaciones Hematológicas de la Academia Nacional de Medicina se observaron: RIPA normal a concentración estándar y ausente a concentración baja, agregación normal con ácido araquidónico y colágeno; ausencia de la segunda onda y de liberación de ATP bajo estimulación con ADP y adrenalina. El contenido de gránulos densos de las plaquetas se halló normal, como se observó mediante la captación de mepacrina. Los resultados se muestran en la figura 1.
Se realizaron VWF:Ag y VWF:RCo, VWF:CB1 y VWF:CB3 y perfil multimérico del VWF. El paciente mostró niveles normales de VWF:Ag, VWF:RCo, patrón multimérico normal, pero bajos niveles de VWF:CB1 y VWF:CB3, con cocientes VWF:CB/VWF:Ag discordantes (<0,7). Se decidió realizar estudios de biología molecular en el exón 28 del gen VWF, no observándose la presencia de VGCEs en dicho exón.
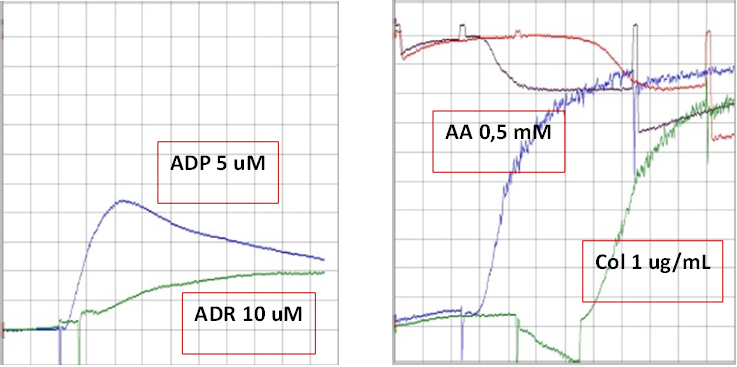
Figura 1. Agregación plaquetaria del paciente con diferentes agonistas
En un tercer estudio se repitieron las determinaciones VWF:Ag, VWF:RCo, que nuevamente se hallaron normales, y VWF:CB1 y VWF:CB3, confirmándose disminuidos. Frente a estos resultados, ante la sospecha de tratarse de VWD2M-C, se decidió realizar la búsqueda de VGCEs en los exones del 29 al 31. Los resultados se muestran en la tabla 1.
Tabla 1. Resultados fenotípicos del paciente
Estudios de laboratorio (rango normal) |
Hospital Italiano |
Dto de Hemostasia y Trombosis, ANM |
|
|
resultados |
1° resultados |
2° resultados |
|
|
RP (150-450x109/L) |
- |
320 |
311 |
|
TC Col/Epi (73-175 seg) |
>300 |
- |
- |
|
TC Col/ADP (50-112 seg) |
150 |
- |
- |
|
TS (1,5-4,5 min) |
- |
4,0 |
- |
|
FVIII (50-150 UI/dL) |
117 |
90 |
110 |
|
VWF:Ag (50-150 UI/dL) |
86 |
69 |
70 |
|
VWF:RCo (50-150 UI/dL) |
- |
55 |
60 |
|
VWF:RCo/VWF:Ag (≥0,7) |
- |
0,8 |
0,9 |
|
VWF:GPIbR (50-150 UI/dL) |
41 |
- |
- |
|
VWF:GPIbR/VWF:Ag (≥0,7) |
0,5 |
- |
- |
|
VWF:CB1 (60-130 UI/dL) |
- |
15 |
18 |
|
VWF:CB1/VWF:Ag (≥0,7) |
- |
0,22 |
0,28 |
|
VWF:CB3 (60-160 UI/dL) |
- |
35 |
42 |
|
VWF:CB3/VWF:Ag (≥0,7) |
- |
0,5 |
0,6 |
|
RIPA 1,2 mg/mL |
- |
Normal |
Normal |
|
RIPA 0,7 mg/mL |
- |
Ausente |
Ausente |
|
VWF perfil multimérico |
- |
Normal |
- |
Mediante los estudios de biología molecular se identificó la VGCE c.5191T>A que codifica para p.Ser1731Thr en el exón 30 en heterocigosis.
Los estudios restantes del paciente no revelaron anomalías.
Conclusión
En nuestro paciente con sintomatología hemorrágica leve-moderada, el hallazgo de los TC prolongados con el PFA-200, VWF:Ag, VWF:RCo normales y VWF:GPlbR ligeramente disminuido pero VWF:CB francamente anormal con ambos colágenos (tipos I y III) despertó la sospecha de VWD2M-C y llevó a la búsqueda de variantes genotípicas en el dominio A3 del VWF y el hallazgo de la mutación p.Ser1731Thr. Esta VGCE está descripta asociada con VWD2M-C en personas con sangrado leve-moderado e inclusive asintomáticas. En nuestro paciente, parecería ser responsable, además, del PFA-200CT anormal, y con ambos cartuchos. Este primer hallazgo suscitó la búsqueda de alteraciones en el dominio A3, con los resultados descriptos.
Sin embargo, no se puede descartar que la alteración de la función plaquetaria con ADP/adrenalina contribuya a la anormalidad del PFA-200CT, particularmente al TC más prolongada con cartucho PFA-COL/Epi que es un patrón más frecuente en las trombocitopatías y en fenotipos con mayor sangrado(9,10).
Dado que el VWF:RCo y GPIbR miden la interacción VWF-plaquetas, y el VWF:CB la interacción VWF-colágeno, es de suma importancia realizar ambos ensayos para evitar la posible falta de diagnóstico de VWW2M-C en pacientes con VWF:Ag ensayos de unión VWF-plaquetas normales.
Bibliografía
1. Ribba AS, Loisel I, Lavergne JM et al. Ser968Thr mutation within the A3 domain of von Willebrand factor (VWF) in two related patients leads to a defective binding of VWF to collagen. Thromb Haemost. 2001;86:848-854
2. Riddell AF, Gomez K, Millar CM et al. Characterization of W1745C and S1783A: 2 novel mutations causing defective collagen binding in the A3 domain of von Willebrand factor. Blood. 2009;114:3489-3496. doi: 10.1182/blood-2008-10-184317
3. Flood VH, Lederman CA, Wren JS et al. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost. 2010;8:1431-1433. doi: 10.1111/j.1538-7836.2010.03869.x
4. Keeling D, Beavis J, Marr R, Sukhu K, Bignell P. A family with type 2M VWD with normal VWF:RCo but reduced VWF:CB and a M1761K mutation in the A3 domain. Haemophilia. 2012;18:e33. doi: 10.1111/j.1365-2516.2011.02676.x
5. Shigekiyo T, Yagi H, Sekimoto E, Shibata H, Ozaki S, Matsumoto M. Identification of a missense mutation (p.Leu1733Pro) in the A3 domain of von Willebrand factor in a family with type 2M von Willebrand disease. Int J Hematol. 2020;111:467-470. doi: 10.1007/s12185-019-02753-4
6. Fels S, Boeckelmann D, Glonnegger H, Büchsel M, Zieger B. Novel likely pathogenic variant in the a3 domain of von Willebrand factor leading to a collagen-binding defect. Hamostaseologie. 2023;43:122-125. doi: 10.1055/a-1701-2181
7. James PD, Connell NT, Ameer Bet al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021;5:280-300. doi:10.1182/bloodadvances.2020003265
8. Woods AI, Sánchez-Luceros A, Kempfer AC, et al. C1272F: a novel type 2A von Willebrand's disease mutation in A1 domain; its clinical significance. Haemophilia. 2012;18:112-116. doi: 10.1111/j.1365-2516.2011.02569.x
9. Favaloro EJ. Clinical utility of closure times using the platelet function analyzer-100/200. Am J Hematol. 2017;92:398-404. doi:10.1002/ajh.24620
10. Guay J, Faraoni D, Bonhomme Fet al. Ability of hemostatic assessment to detect bleeding disorders and to predict abnormal surgical blood loss in children: a systematic review and metaanalysis. Paediatr Anaesth. 2015;25:1216-26. doi:10.1111/pan.12723