Síndrome de Kilt: causa infrecuente de trombosis ileofemoral bilateral. Reporte de caso
Kilt syndrome: uncommon cause of bilateral ileofemoral thrombosis. Case report
Cavia S1; Wolhein DS1; Bernard H1; Chávez L1; Benavídez N1; Beligoy M1; Devecchi A1; Marull M1; Fernández C1; Ventos R 2
1- Servicio de Hematología - Hospital Escuela Dr. Ramón Madariaga. Posadas, Misiones. Argentina.
2- Servicio de Clínica Médica - Hospital Escuela Dr. Ramón Madariaga. Posadas, Misiones. Argentina.
https://orcid.org/0009-0008-8656-4187 Sergio Cavia
https://orcid.org/0000-0002-6669-1970 Wolhein Diego
https://orcid.org/0009-0001-1581-0706 Marull Mariana
https://orcid.org/0009-0009-8388-0536 Chávez Luciana
https://orcid.org/0009-0005-7660-1990 Nadia Benavídez
https://orcid.org/0009-0006-2509-7787 Haydée Bernard
https://orcid.org/0009-0004-9196-2623 Devecchi Amalia
https://orcid.org/0009-0007-0885-4069 Itamar Cruz
https://orcid.org/0009-0007-3440-7967 Ventos Romina
https://orcid.org/0009-0008-6615-3364 Fernández Claudia
Palabras claves: de Kilt,
trombosis bilateral,
agenesia vena cava.
Keywords: Kilt syndrome,
bilateral thrombosis,
cava vein agenesis.
Resumen
La ausencia de la vena cava inferior es una afección poco común, descubierta usualmente de forma casual. Se describe en la literatura actual como un factor de riesgo poco frecuente de TVP. La presencia de anomalías renales y de la VCI en los estudios por imágenes junto con trombosis de las extremidades inferiores se conoce colectivamente como síndrome de Kilt (aunque las anomalías renales no son excluyentes de dicho síndrome). Presentamos un caso de síndrome de Kilt, diagnosticado por primera vez a los 32 años de edad, en contexto de dolor intensidad 10/10 en miembro inferior izquierdo asociado a edema y asimetría.
Summary
The absence of the inferior cava vein is a rare condition, usually discovered by chance. It is described in the current literature as a rare risk factor for DVT. The presence of renal and ICV abnormalities on imaging studies along with lower extremity thrombosis is collectively known as Kilt syndrome (although renal abnormalities are not exclusive of Kilt syndrome). We present a case of Kilt syndrome, diagnosed for the first time at 32 years of age, in the context of pain intensity 10/10 in the left lower limb associated with edema and asymmetry.
Material y método: Descripción de un caso clínico y revisión de la literatura.
Caso clínico
Paciente de 32 años de edad de sexo masculino, sin antecedentes patológicos de importancia, que refiere iniciar en el mes de abril del 2024 con dolor intensidad 10/10, edema, eritema e impotencia funcional del miembro inferior derecho, realizando únicamente tratamiento sintomático con analgésicos tipo AINES. Por progresión de los síntomas 2 semanas después con afectación del miembro contralateral decide consultar a servicio de emergencia de nuestro nosocomio realizándose al ingreso ecodoppler venoso bilateral, constatándose trombosis femoral bilateral.
El examen físico revela aumento de diámetro y temperatura de ambos miembros inferiores.
Se decide instauración de tratamiento con enoxaparina a 1 mg/kg cada 12 h, sin presentar mejoría a las 72 h, con respecto al dolor y edema, por lo que se decide la realización de una angiotomografía de abdomen y pelvis con contraste, constatándose, además del compromiso profundo de ambas venas ilíacas, la malformación anatómica de la vena cava inferior e informándose como agenesia de la misma (Figura 1).
Al solicitarle al paciente estudios previos, se comprueba que en una resonancia de columna por antecedente de hernia de disco ya se informaba la malformación anatómica.
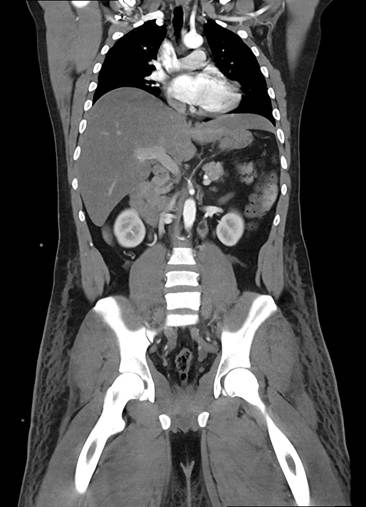
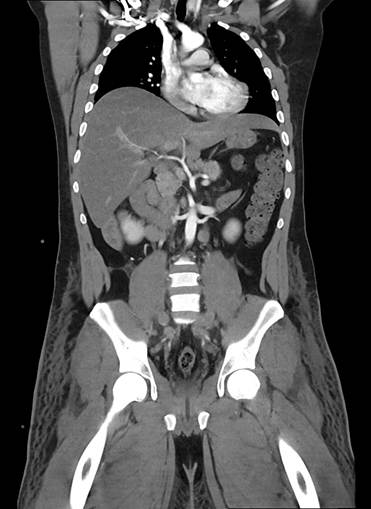
Figura 1. Tac de abdomen (corte coronal). Ambas venas ilíacas dilatadas tortuosas heterogéneas con defecto de relleno post contraste vena cava inferior colapsada, se asocia además de tejido celular subcutáneo. Riñones de forma, tamaño y situación normales sin alteraciones densitometrías. Hallazgos vinculables a trombosis de ambas venas ilíacas y agenesia de vena cava inferior.
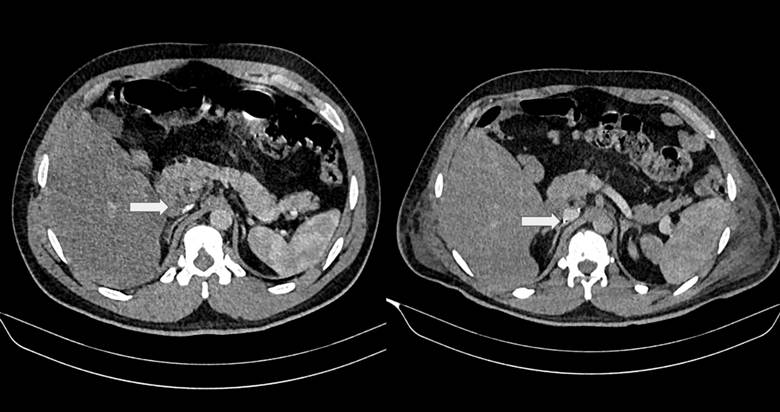
Figura 2. Tac de abdomen (corte axial). Se logra apreciar la malformación a nivel infrarrenal de la vena cava inferior con colapso.
Con dichos hallazgos y ante la sospecha de síndrome de Kilt, se decide interconsultar con el servicio de Hemodinamia para evaluación y tratamiento.
Se realiza posteriormente angiografía, confirmándose la agenesia de vena cava inferior infrarrenal con colapso superior por lo que se decide en un primer momento la colocación de un filtro de vena cava retrohepático. Posteriormente, a la semana, se decide nueva intervención para infusión de trombolíticos rTPA (10 mg de alteplasa en bolo, correspondientes a 5.800.000 UI) con posterior colocación de catéter para infusión continua ,90 mg como perfusión intravenosa a velocidad constante durante 2 horas hasta una dosis máxima total de 100 mg.
A las 24 h, encontrándose en unidad de coronaria, se controla APTT y fibrinógeno en rango, por lo que se inicia bomba de infusión de heparina manteniendo el APPT en rango terapéutico entre 1.5 y 2.5 del valor superior normal.
Presenta franca mejoría clínica y control sintomático, por lo que se decide rotar a enoxaparina y cabalgamiento con anticumarínicos.
Actualmente se encuentra en seguimiento ambulatorio con buena evolución.
Durante la internación se realizaron además los siguientes exámenes complementarios:
Ecodoppler cardíaco: dentro de parámetros normales (para descartar otras malformaciones anatómicas).
Estudios de trombofilia (factor V de Leyden y mutación protombina G20210A): normales.
JAK2 - citometría de flujo para HPN: sin hallazgos patológicos.
RNM de columna lumbar (previo a la internación): se observa dilatación de estructura de aspecto vascular venoso epidural en el segmento lumbar bajo, asociada a múltiples imágenes compatibles con vasos dilatados e irregulares en la región retroperitoneal del segmento comprendido entre L4 y L5, con dilatación de estructuras venosas en espacios paravertebrales. Este hallazgo podría estar relacionado a congestión y circulación colateral secundaria a agenesia de vena cava inferior.
Discusión
El síndrome de Kilt es extremadamente raro, presentándose en pacientes de sexo masculino, jóvenes, con TVP ileofemoral bilateral extensa, asociada a agenesia de vena cava inferior, pudiendo coexistir o no con malformaciones renales(1).
Las anomalías de la vena cava inferior en sí mismas son raras y ocurren en aproximadamente en el 0,6 al 2 % de los individuos con cardiopatía estructural congénita, pero sólo en el 0,3 al 0,6 % de la población general.
Las anomalías más comunes de la VCI son la VCI doble, la VCI del lado izquierdo, la agenesia o ausencia de la VCI y la vena renal izquierda retroaórtica. En pacientes jóvenes con TVP, se estima que existe una tasa más alta de anomalías de la VCI que en la población general, es decir, un 5% en comparación con el 0,5% esperado(4).
La ausencia de algún segmento de una VCI madura a menudo se diagnostica de manera incidental, más comúnmente durante la evaluación de la TVP (como en nuestro caso) o en otras imágenes transversales de rutina del abdomen y la pelvis.
Se reconoce que la ausencia de VCI está asociada con una TVP idiopática. La TVP en presencia de una VCI ausente se presenta con mayor frecuencia en varones jóvenes y, generalmente, en ausencia de factores desencadenantes.
Las anomalías de la VCI son consecuencia de la persistencia o regresión anormal de las venas embriológicas. La ausencia de toda la VCI es de origen embriológico, mientras que la ausencia de sólo la VCI infrarrenal, como en el caso presentado, probablemente se deba a una trombosis intrauterina o perinatal(2).
En el contexto clínico correcto, las anomalías de la VCI y el síndrome de Kilt deben considerarse en pacientes que presentan TVP bilaterales proximales, extensas y recurrentes y en aquéllos con insuficiencia venosa de aparición temprana(3).
La embriogénesis de la VCI es compleja. La VCI normal se compone de cuatro segmentos: hepático, suprarrenal, renal e infrarrenal. El segmento hepático deriva de la vena vitelina. La vena subcardinal derecha se desarrolla en el segmento suprarrenal mediante la formación de la anastomosis subcardinal-hepática. El segmento renal se desarrolla a partir de las anastomosis suprasubcardinal y postsubcardinal derechas. Se acepta generalmente que el segmento infrarrenal deriva de la vena supracardinal derecha. En la región torácica, las venas supracardinales dan lugar a las venas ácigos y hemiácigos. En el abdomen, las venas postalinales son reemplazadas progresivamente por las venas subcardinales y supracardinales, pero persisten en la pelvis como las venas ilíacas comunes(5). Algunos autores proponen que la ausencia de la VCI puede ser el resultado de una trombosis perinatal o de una trombosis intrauterina, con obliteración y posterior reabsorción(6). Sin un desarrollo normal de la VCI infrarrenal, las venas iliofemorales drenan en las venas ácigos y hemiácigos a través de colaterales paravertebrales anteriores. Como todos los vasos colaterales son mucho más pequeños en calibre con respecto a la VCI normal, es comprensible que dichas vías colaterales puedan conducir a estasis venosa crónica y trombosis de la extremidad inferior(4).
La agenesia de la VCI infrarrenal es una anomalía rara.
Los pacientes con agenesia de la VCI pueden presentar un cuadro clínico variado. Algunos pueden ser asintomáticos y la ausencia de la VCI se encuentra como un hallazgo incidental; otros pueden presentarse con trombosis venosa y sus consecuencias, como en nuestro caso(5).
No ha habido un consenso claro en la literatura sobre el manejo de los pacientes con agenesia de la VCI y trombosis venosa. La mayoría de los pacientes descritos han sido tratados con anticoagulación y medias elásticas. Pocos han requerido intervención quirúrgica para el alivio de los síntomas. La duración del tratamiento con anticoagulación tampoco ha sido bien descrita en la literatura. A diferencia de los factores de riesgo adquiridos, que pueden ser corregibles, los pacientes con el factor de riesgo de ausencia de VCI infrarrenal pueden tener un riesgo de tromboembolia de por vida. Se necesitan más estudios de seguimiento de estos pacientes para determinar la recurrencia de los síntomas, la duración del tratamiento, las opciones alternativas de tratamiento y el pronóstico(4).
Bibliografía
1. Onbaş O, Kantarci M, Koplay M, Olgun H, Alper F, Aydinli B, Zirek H, Ceviz N. Congenital anomalies of the aorta and vena cava: 16-detector-row CT imaging findings. Diagn Interv Radiol. 2008 Sep;14(3):163-71.
2. Li SJ, Lee J, Hall J, Sutherland TR. The inferior vena cava: anatomical variants and acquired pathologies. Insights Imaging. 2021 Aug 30;12(1):123. doi: 10.1186/s13244-021-01066-7.
3. Droog W, van Beek AJ, Kooijman R. An extraordinary cause for deep venous thrombosis. BMJ Case Rep. 2011 Feb 9;2011:bcr0120102695. doi: 10.1136/bcr.01.2010.2695.
4. Bami S, Vazquez Y, Chorny V, Goldfisher R, Amodio J. Deep Venous Thrombosis of the Leg, Associated with Agenesis of the Infrarenal Inferior Vena Cava and Hypoplastic Left Kidney (KILT Syndrome) in a 14-Year-Old Child. Case Rep Pediatr. 2015;2015:864047. doi: 10.1155/2015/864047.
5. Bassa BA, Ryan D, Reid E, Bolster F, Breslin T. A rare case of KILT syndrome in Ireland: A case report. Thrombosis Update. 2023;10:100131. doi.org/10.1016/j.tru.2023.100131. (https://www.sciencedirect.com/science/article/pii/S2666572723000020)
6. Castro FJ, Pérez C, Narváez FJ, Gacía A, Biosca M, Vilaseca J, Vives J, Argiles JM. Agenesia de vena cava inferior como factor de riesgo de tromboembolismo pulmonar [Congenital absence of the inferior vena cava as a risk factor for pulmonar thromboembolism]. An Med Interna. 2003 Jun;20(6):304-6.