Crisis blástica de células dendríticas plasmocitoides y crisis blástica megacarioblástica en dos pacientes con leucemia mieloide crónica: reporte de casos.
Plasmocytoid dendritic cell blast crisis and megakaryoblastic blast crisis in two patients with chronic myeloid leukemia: case report
D’Antuoni, J; Tito, L; Tellas, M; Freilich, F; Cortez, J; Reina, J; Márquez, N; Ross, S; Torres, A; Quispe, D; Bertolino,Y; Mazzeo, M; Rey, I.
D'antuoni Julieta https://orcid.org/0009-0002-2079-2482
Rey, Irene https://orcid.org/0000-0003-3403-5124
Freilich, Franco: https://orcid.org/0009-0004-8017-6627
Torres, Adriana https://orcid.org/0009-0009-9158-5326
Ross. Josefina Solange https://orcid.org/0009-0008-5794-4332
Bertolino, Yamila https://orcid.org/0009-0007-7996-5639
Jenifer Lorena Reina Noboa https://orcid.org/0000-0002-7779-4203
Javier Cortez Velasquez https://orcid.org/0009-0009-0736-0715
Noelia Márquez Valdivia https://orcid.org/0009-0003-2971-8980
Luis Alberto Tito Quispe https://orcid.org/0009-0004-1893-9638
Daniel Quispe Mamani https://orcid.org/0009-0005-9874-9478
Maria Aurelia Mazzeo https://orcid.org/0009-0001-2287-2032
Micaela Tellas https://orcid.org/0009-0004-8541-0144
Hospital General de Agudos José María Ramos Mejía. CABA, Argentina.
Palabras clave: leucemia mieloide crónica,
crisis blástica,
células dendríticas plasmocitoides,
megacarioblástica.
Keywords: chronic myeloid leukemia,
blast crisis,
plasmacytoid dendritic cells,
megakaryoblastic.
Resumen
La crisis blástica (CB) sigue siendo el principal desafío de la leucemia mieloide crónica (LMC). El tratamiento incluye un inhibidor de tirosina quinasa (ITK) asociado o no a poliquimioterapia, seguido de trasplante de células madre hematopoyéticas (TCPH) en pacientes aptos. El fenotipo leucémico incluye tanto estirpe mieloide como linfoide. La CB mieloide con diferenciación megacarioblástica es infrecuente, mientras que la diferenciación a células dendríticas plasmocitoides es extremadamente rara en la literatura. A continuación, presentamos un paciente con LMC en crisis blástica megacarioblástica (CB-MGK) y un caso de crisis blástica de células dendríticas plasmocitoides (CB-CDP), que recibieron tratamiento combinado de ITK y poliquimioterapia con excelentes resultados pese al mal pronóstico.
Abstract
Blastic crisis (BC) remains the main challenge in chronic myeloid leukemia (CML). Treatment typically involves a tyrosine kinase inhibitor (TKI) alone or in combination with polychemotherapy, followed by hematopoietic stem cell transplantation (HSCT) for eligible patients. The leukemic phenotype includes both myeloid and lymphoid lineages. Myeloid BC with megakaryocytic differentiation is rare, while differentiation to plasmacytoid dendritic cells is extremely uncommon in the literature. We present a patient with CML in megakaryoblastic blastic crisis (BC-MGK) and a case of blastic crisis with plasmacytoid dendritic cells (BC-PDC), both of whom received combined treatments with TKI and polychemotherapy with excellent outcomes despite the poor prognosis.
Introducción
La LMC pertenece al grupo de las neoplasias mieloproliferativas crónicas, y se caracteriza por la presencia de la translocación recíproca t (9;22) (q34.1; q11.2), que resulta en la formación del cromosoma Filadelfia, el cual contiene el gen de fusión BCR::ABL1 cuya actividad de tirosina quinasa constitutiva desregula vías de transducción de señales llevando a la proliferación celular sin arresto madurativo(1). Los pacientes pueden presentarse al diagnóstico en tres diferentes fases: fase crónica (FC), fase acelerada (FA) o CB, que representan la evolución natural de la enfermedad(1). Aunque el curso trifásico de la LMC ha sido históricamente bien reconocido en la literatura y la comunidad médica, la definición precisa es aún motivo de discusión y puede variar considerablemente (la OMS ha modificado en su última revisión la definición de fase acelerada)(1,2). La introducción de los ITK en el año 2001, con la aprobación del imatinib, ha minimizado la posibilidad de progresión de la LMC a CB(3). Sin embargo, continúa siendo un desafío terapéutico, con un pronóstico sombrío a corto plazo, siendo una enfermedad rápidamente fatal. Presentamos dos formas infrecuentes de LMC en CB, una con diferenciación megacarioblástica (caso 1) y otra a células dendríticas plasmocitoides (caso 2).
Caso clínico N° 1
Paciente masculino de 44 años, con diagnóstico en diciembre del 2022 en su provincia natal (Chubut, Argentina) de LMC en FC en contexto de astenia, adinamia, pérdida de peso y distensión abdominal, esplenomegalia de 10 cm bajo reborde costal izquierdo, SOKAL alto riesgo y ELTS alto riesgo. Inicia tratamiento con dasatinib 100 mg/día. A los 6 meses presenta respuesta molecular nula (RMN). A la espera de un ITK de segunda línea desarrolla pancitopenia, y suspende tratamiento con dasatinib. Inicia mantenimiento con eritropoyetina (EPO) y factores estimulantes de colonias granulocíticas (GCSF) en espera de entrega de ITK. En el mes de noviembre del 2023 evoluciona con un síndrome febril a foco probable en mucosa oral, en contexto de neutropenia febril. Es internado en un hospital zonal de Chubut e inicia tratamiento antibiótico empírico, suspende EPO y GCSF.
Por la presencia en el frotis de sangre periférica de células atípicas mayor al 50% compatibles con blastos, se solicita derivación a hospital de mayor complejidad por sospecha de progresión a CB. Es admitido en el Servicio de Hematología del Hospital General de Agudos J. M. Ramos Mejía, vía derivación por avión sanitario en el mes de diciembre del 2023.
A su ingreso presenta al examen físico petequias en tórax superior, dorso, miembros superiores e inferiores, esplenomegalia infraumbilical mayor a 10 cm, hepatomegalia 8 cm, lesiones tipo vesículas en ramillete en labio inferior y superior y mal estado bucodental. En la tomografía computada se informa: esplenomegalia masiva 350 x 200 mm que desplaza órganos abdominales. Hepatomegalia 250 x 200 mm de diámetro.
Figura 1, A y B
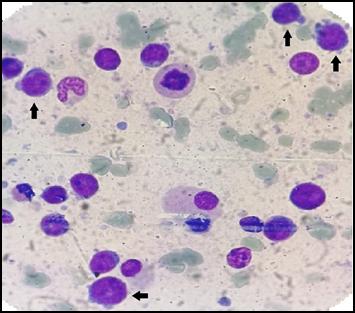
Laboratorio de ingreso: Hto 21.8%, Hb 7.1 g/dL, GB 4030/mm3 (neutrófilos 24%, linfocitos: 36%, monocitos: 2%, eosinófilos: 0%; blastos 38%), plaquetas: 14000/mm3, LDH: 644 UI. Función renal y hepática sin alteraciones, sin evidencia de síndrome de lisis tumoral. En el frotis de sangre periférica se observa 38% de células gran tamaño, alta relación núcleo citoplasma, con nucleolos evidentes, prolongaciones citoplasmáticas (blebs de membrana y citoplasma basófilo) y ausencia de gránulos (Figura 1, A y B).
Se realiza una punción/biopsia de médula ósea (MO), con evidencia de infiltración de células de gran tamaño, similares a las evidenciadas en el frotis de sangre periférica, que representan el 68% de la celularidad en el extendido de MO (Figura 2, A y B). La citometría de flujo (CMF) informa 56.7 % blastos mieloides con diferenciación a linaje de megacariocito: células de gran tamaño, CD45+, CD34++, MPO (-), CD79a (-), CD19(-), CD7++, CD3s(-), CD3ic(-), CD117(-), HLA-DR(-), CD16(-), CD13+, CD11b++, CD10(-), CD64(-), CD35+, CD14(-), Irem-2(-), CD36++, CD105+, CD33++, CD71+, CD15(-), CD123(-), CD56-/+(3%), CD38++, CD61&CD41+, CD25(-), CD9(-), CD203c+. Conclusiones: Presencia de blastos mieloides con signos de diferenciación a linaje megacariocítico en rango leucémico. Hallazgos inmunofenotípicos compatibles con leucemia megacarioblástica aguda.
Figura 2 A, B.
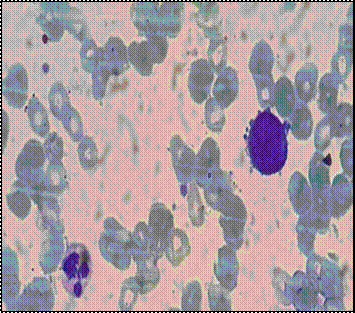
En la anatomía patológica se describe una ocupación de los espacios medulares en un 100% por celularidad hematopoyética, que desplaza al tejido adiposo obliterando los espacios medulares. Los precursores CD34+ representan un 80% de la celularidad. Conclusión: hallazgos morfológicos compatibles con LMC en CB asociada a fibrosis reticulínica grado 1.
Se realiza también el estudio citogenético, que informa 46 XY t (9;22) (q32;q11.2) [20 metafases], biología molecular para BCR::ABL: p210 detectable y mutación T315I: negativa. Se diagnostica LMC en crisis blástica megacarioblástica.
En la valoración integral se considera al paciente apto para poliquimioterapia, e inicia tratamiento con esquema 7+3 (citarabina 100 mg/m2 e idarrubicina 12 mg/m2) asociado a ponatinib 45 mg/día.
Debido a pancitopenia prolongada por 6 semanas con sostén transfusional con hemoderivados por trombocitopenia grado 4 de difícil manejo con manifestaciones de sangrado mucocutáneo, anemia sintomática y episodios de neutropenia febril, suspende el ITK en múltiples oportunidades realizando un tratamiento intermitente durante las primeras semanas de tratamiento. Por un cuadro clínico de neumonía con compromiso nodular pulmonar bilateral debido a una infección fúngica invasora (IFI) con curva de galactomananos positiva en sangre (probable aspergilosis pulmonar) se realiza un lavado broncoalveolar en Hospital Muñiz (sin aislamiento bacteriano o fúngico) e inicia tratamiento antifúngico durante 3 meses con voriconazol y posterior profilaxis.
Durante el tratamiento conjunto con ponatinib y voriconazol (drogas con potencial interacción) presenta hepatotoxicidad grado 3, que requiere suspensión del ponatinib por dos semanas con descenso de dosis a 15 mg/día. Se cambia tratamiento de voriconazol a posaconazol, sin nuevas complicaciones.
El paciente logra la remisión completa (RC) por CMF a las 6 semanas de iniciado el tratamiento, y se encuentra a la espera de TCPH con donante histoidéntico no relacionado. En tratamiento con ponatinib 15 mg/día. Presenta EMR negativa, con una respuesta molecular completa (RMC) de 4.5 por el estándar internacional (EI) a los 6 meses de iniciado el ITK.
Caso clínico N° 2
Paciente masculino de 34 años. Inicia en mayo 2022 con dolor en hipocondrio izquierdo, hiporexia no selectiva, pérdida de peso y epistaxis intermitentes autolimitadas. En junio de 2022 evoluciona con registros febriles y tos persistente, por lo que acude al hospital Muñiz. Es valorado en contexto de neumonía de la comunidad, se descarta Covid-19 por PCR e inicia tratamiento antibiótico empírico con buena evolución. Durante valoración clínica se realiza laboratorio, donde se evidencia bicitopenia y leucocitosis: Hto 24.8%, Hb 7.7 g/dL, VCM 94 fl, GB 344070/mm3 y plaquetas 110000/mm3. En el frotis de sangre periférica se destaca la presencia de desviación a la izquierda con 40% de células de alta relación núcleo citoplasma y cromatina laxa, con nucleolos evidentes. Se decide derivación al Hospital General de Agudos J. M. Ramos Mejía por sospecha de CB de LMC de novo.
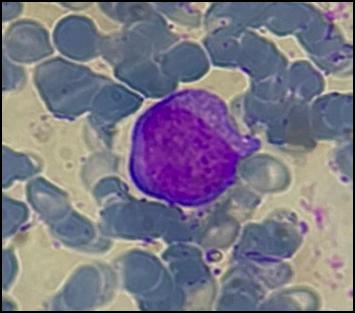
A fines de junio del 2022 se realiza aspiración y biopsia de MO. En el extendido de médula ósea presenta infiltración de células de gran tamaño, con alta relación núcleo citoplasma y 2 a 4 nucleolos evidentes en rango leucémico (Figura 3 A, B.) asociado a hiperplasia de la serie eritroide. La CMF informa el siguiente resultado: población de blastos de mediano tamaño, mediana complejidad interna que representa un 37.9 % del total celular. Esta población presenta el siguiente inmunofenotipo: CD45+, CD34++, MPO(-), CD79a(-), CD19(-), CD7++, CD3s(-), CD3ic(-), CD117-/+, HLA-DR++, CD16(-), CD13-/+, CD11b-/+, CD10(-), CD64(-), CD35(-), CD14(-), Irem-2(-), CD36-/+, CD105(-), CD33++, CD71+, CD15(-), CD123++, CD56-/+, CD38-/++, CD4-/+, NG2(-), CD304(-), CD2-/+. Nota: Los blastos no presentan una clara comisión de linaje.
Figura 3 A y B.
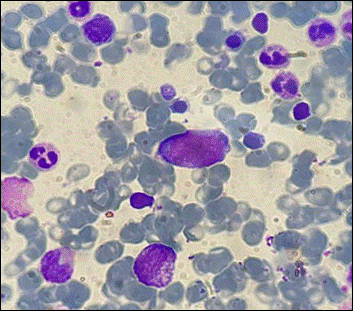
Si bien la expresión intensa de CD33 no es habitual en precursores de célula dendrítica, la expresión de los marcadores CD36, CD4 y CD56 (aunque es parcial) junto con la expresión intensa de HLA-DR favorece una diferenciación hacia este linaje celular. Presencia de blastos en rango leucémico (37.9%), hallazgos inmunofenotípicos compatibles con leucemia aguda con diferenciación a células dendríticas plasmocitoides. Se realiza estudio citogenético 46;XY.t(9;22)(q32;q11.2) [20 metafases], biología molecular para BCR::ABL1 p210 detectable. Anatomía patológica de médula ósea: infiltración de médula ósea por leucemia aguda.
Por presentar diferenciación a células dendríticas se realiza valoración del LCR por CMF, y se identifica una población de blastos de tamaño mediano, complejidad interna mediana que representa un 14.5 % del total celular. Esta población presenta un inmunofenotipo similar al evidenciable en MO. Se define diagnóstico de LMC en crisis blástica de células dendríticas plasmocitoides con compromiso del sistema nervioso central.
Por leucocitosis inicia citorreducción con 3 gramos/día de hidroxiurea durante 13 días, y luego de una valoración multidisciplinaria se lo considera apto para poliquimioterapia e inicia protocolo HyperCVAD a principios de julio del 2022 + dasatinib 140 mg (por citopenias, se reduce dosis a 100 mg/día). Recibe en total 9 punciones lumbares con quimioterapia intratecal y posterior negativización del compromiso del SNC durante la inducción.
Presenta complicaciones infectológicas asociadas a neutropenia febril con buena evolución clínica luego del tratamiento antibiótico en cada ciclo de quimioterapia. Finalizado el C4 del protocolo HyperCVAD, se realiza valoración de enfermedad de base, constatándose EMR positiva en MO del 0,1% en diciembre del 2022 con SNC negativo. Se decide modificar esquema quimioterápico a FLAGIDA (fludarabina, citarabina e idarrubicina) asociado a ponatinib 45 mg/día y quimioterapia intratecal profiláctica en enero del 2023. Presenta citopenias prolongadas y neutropenia febril con tratamiento antibiótico empírico y buena evolución, presentando al día 35 de quimioterapia en febrero del 2023 nueva valoración de enfermedad con EMR negativa en MO y en SNC, logrando la RC. Mantiene RMC 4.0 a los 6 y 9 meses. Se solicita TCPH, manteniendo tratamiento con ponatinib 45 mg/día, se destacan demoras en realización del TCPH debido a no contar con un donante relacionado y a la baja de un donante internacional israelí. El 06/02/2024, luego de un año de haber logrado RC, se realiza la infusión de CPH en centro de trasplante de la provincia de Buenos Aires con donante no relacionado. Actualmente continua seguimiento post trasplante en buen estado general, con EMR negativa y una RMC 4.5 por el EI a los 6 meses del TCPH.
Discusión
La presencia de más de 20% de blastos en sangre periférica, médula ósea o un cluster de blastos en la biopsia de MO o una masa extramedular, en el contexto de LMC definen la crisis blástica(1,4). El fenotipo más comúnmente visto en la crisis blástica es el linaje mieloide (60%) seguido de linfoide y eritroide, mientras que la CB-MGK representa menos del 3% y de la forma CB-CDP sólo existen reportes de casos en la literatura(4). Ambos inmunofenotipos pueden representar un desafío diagnóstico y terapéutico por la escasa información bibliográfica, en especial en ausencia de diagnóstico previo de LMC en FC.
La leucemia mieloide aguda (LMA) con diferenciación megacariocítica (LMKA) representa menos del 5% de todas las LMA y se define como una leucemia con al menos un 20% de blastos, de los cuales ≥50% son de linaje megacariocítico(5). Esta leucemia tiene una distribución por edad binodal con picos en niños menores de 3 años y en adultos mayores(6). Muchos casos pediátricos de LMKA están asociados con el síndrome de Down (trisomía 21), que se clasifica como una entidad separada en la clasificación de la Organización Mundial de la Salud (OMS): leucemia mieloide asociada con el síndrome de Down(4). La fibrosis de médula ósea es un hallazgo muy frecuente y común en los casos conocidos de este tipo de leucemia(7-9), coincidente con nuestro caso clínico.
La crisis blástica megacariocítica es extremadamente infrecuente y existe dificultad al momento del diagnóstico para diferenciarla de la LMKA de novo, sobre todo en los pacientes en los que no se dispone de antecedentes de LMC en fase crónica(8). La característica distintiva entre ambos grupos es la presencia del transcripto BCR/ABL p210 y la expresión de antígenos específicos de plaquetas CD41 y CD61 en la superficie de los blastos en pacientes con LMC(8). La morfología de los megacarioblastos presenta una alta relación núcleo-citoplasma, núcleo irregular con presencia de 1 a 3 nucléolos, citoplasma basófilo con vacuolas y en algunos casos formación de pseudópodos o prolongaciones citoplasmáticas(9). En la CMF los blastos muestran positividad para uno o más de los antígenos megacarioblásticos CD41, CD61 o CD42(9). La presencia de esplenomegalia, basofilia y trombocitosis están relacionados con el antecedente de LMC. El análisis morfológico e inmunofenotípico de nuestro paciente reveló la presencia de una única población de blastos leucémicos megacarioblásticos (LMA M7) asociada con el antecedente de LMC. En la literatura existen reportes de casos de pacientes con LMC en CB-MGK(10-17). Aunque en términos generales el pronóstico es desfavorable, se pueden observar algunos reportes de casos de paciente que han alcanzado la remisión completa luego de la combinación de poliquimioterapia con ITK, como se observa en los casos publicados por Pelloso, Wu y Rashi(11,12,17).
Las células dendríticas plasmocitoides forman parte del sistema inmune innato, y sus formas malignas son reconocidas como neoplasias de células dendríticas plasmocitoides blásticas por la WHO(1). Las CB-CDP representan el 0.44% de las patologías oncohematológicas, con una edad media de 53 años y un predominio 2-4:1 para los hombres, con dos picos binodales (mayores de 60 años y menores de 20 años)(18). Clínicamente se presenta como una patología agresiva y de difícil manejo. El diagnóstico se suele realizar en contexto clínico de pancitopenia o lesiones sugestivas en piel. Ante la sospecha debe realizarse evaluación dermatológica y biopsia de lesiones en piel (si existiesen), PET TC corporal total (en ausencia de células dendríticas en sangre periférica o médula ósea), biopsia de médula ósea con CMF para determinar fenotipo, estudio citogenético y NGS (si se encuentra disponible)(18). Aquellos pacientes con síntomas neurológicos deben ser evaluados con imágenes y punción lumbar(18). Suele manifestarse con compromiso de piel (80%), médula ósea, órganos linfáticos, SNC y líquido cefalorraquídeo(19). Más de un 60% de los pacientes tiene compromiso de médula ósea al diagnóstico. Habitualmente expresan CD4 y CD56, siendo positivos para al menos uno de los marcadores adicionales de células dendríticas, como son el CD123, TCF4, TCL1, CD304 y CD303(19). Sin embargo, la mayor parte de la información disponible en la bibliografía corresponde a pacientes con CB-CDP, sin relación con LMC. Considerada una patología extremadamente infrecuente, el hallazgo de la translocación t(9;22) (q34;q11.2) y el transcripto BCR::ABL1, en contexto de una CB-CDP sólo presenta reportes de casos en la literatura(20-21). Es infrecuente que esta patología se presente sin lesiones en la piel cuando se presenta como una leucemia de novo(19), en nuestro caso, el paciente no presentaba lesiones dérmicas.
El tratamiento de la LMC-CB, de novo o por progresión de una FC, debe incluir esquemas de poliquimioterapia según el linaje (mieloide, linfoide, ambiguo) y un ITK de segunda o tercera generación, seguido de un trasplante alogénico de médula ósea con el mejor donante posible(22). En caso de no ser candidato a trasplante de médula ósea, el uso de ITK en monodroga minimizando los efectos adversos de la poliquimioterapia debe ser considerado, como se muestra en el registro del manejo de la fase blástica de la European Leukemia Net (ELN) presentado en el 2024, con 21,1% de pacientes tratados con ITK solo para un n de 240 pacientes, aunque los resultados son considerablemente más pobres(23). La sobrevida global en pacientes con LMC-CB según la ELN es de 23,8 meses. Siempre debe tenerse como opción el ingreso de los pacientes a un ensayo clínico, dado que la sobrevida de aquellos pacientes que no alcanzan el trasplante alogénico de médula ósea es menor a un año. El uso de esquemas de poliquimioterapia con citarabina y antraciclinas +/- fludarabina (esquema 7+3, FLANG, FLAGIDA) asociados a un ITK como dasatinib o ponatinib son los más difundidos en la literatura, con tasas de remisión completa entre un 30 y un 60%(24). Dado que tanto la LMC en CB-MGK como en CB-CDP son de estirpe mieloide, la extrapolación de estos esquemas es una opción viable ante la falta de bibliografía, quedando reflejado en los buenos resultados obtenidos con los pacientes presentados.
Conclusión
La progresión a CB continúa siendo un desafío terapéutico en los pacientes con LMC, incluyendo tanto pacientes resistentes al tratamiento con ITK como aquéllos diagnosticados en fase blástica de novo. En este caso, presentamos a dos pacientes con dos formas de CB extremadamente infrecuentes, lo cual suma un desafío diagnóstico y terapéutico tanto por la falta de consensos en cuanto al tratamiento óptimo, como por el desconocimiento general de la fisiopatología y las características biológicas de estas dos presentaciones. El diagnóstico correcto debe incluir tanto el aspirado de médula ósea como la biopsia con anatomía patológica de médula ósea, dada la alta incidencia de aspirados secos por la fibrosis medular, que es un factor de pronóstico adverso perse.
La falta de literatura a nivel mundial dificulta considerablemente la elección adecuada del tratamiento exitoso de pacientes con LMC en CB-MGK y CB-CDP. La utilización de esquemas de poliquimioterapia con citarabina asociados al uso de un ITK de segunda o tercera generación podría tener los mejores resultados. En este reporte presentamos dos pacientes con un tratamiento exitoso de esquemas combinados de poliquimioterapia y ponatinib, incluso con dosis reducidas para evitar la toxicidad hematológica y sistémica por los ITK. El trasplante alogénico de médula ósea sigue siendo la única opción para lograr una sobrevida global prolongada en aquellos pacientes aptos.
Resulta imprescindible dar conocimiento de aquellos casos clínicos que se presenten para enriquecer las estrategias diagnósticas, de seguimiento y de tratamiento para aquellos profesionales que se topen con el reto de lograr un éxito terapéutico en una patología de mal pronóstico.
Bibliografía
1. Khoury JD, Solary E, Abla O y col. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022 Jul;36(7):1703-1719. doi: 10.1038/s41375-022-01613-1.
2. Baccarani M, Deininger MW, Rosti G y col. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122:872-884.
3. Hochhaus A, Baccarani M, Silver RT y col. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020 Apr;34(4):966-984. doi: 10.1038/s41375-020-0776-2.
4. Arber DA, Orazi A, Hasserjian R y col. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-2405.
5. Arber DA, Brunning RD, Orazi A, y col. Acute myeloid leukaemia, NOS. In: Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2017:156-168.
6. Giri S, Pathak R, Prouet P, Li B, Martin MG. Acute megakaryocytic leukemia is associated with worse outcomes than other types of acute myeloid leukemia. Blood. 2014;124(25):3833-3834.
7. Teyssier AC, Lapillonne H, Pasquet M y col. Acute megakaryoblastic leukemia (excluding Down syndrome) remains an acute myeloid subgroup with inferior outcome in the French ELAM02 trial. Pediatr Hematol Oncol. 2017;34(8):425-427.
8. Pullarkat ST, Vardiman JW, Slovak ML y col. Megakaryocytic blast crisis as a presenting manifestation of chronic myeloid leukemia. Leuk Res. 2008 Nov;32(11):1770-5. doi: 10.1016/j.leukres.2008.02.025.
9. Chisholm KM, Smith J, Heerema-McKenney AE y col. Pathologic, cytogenetic, and molecular features of acute myeloid leukemia with megakaryocytic differentiation: A report from the Children's Oncology Group. Pediatr Blood Cancer. 2023 May;70(5):e30251. doi: 10.1002/pbc.30251.
10. Bryant BJ, Alperin JB, Elghetany MT. Paraplegia as the presenting manifestation of extramedullary megakaryoblastic transformation of previously undiagnosed chronic myelogenous leukemia. Am J Hematol. 2007;82:150-4.
11. Pelloso LAF, Baiocchi OCG, Chauffaille MLLF y col. Megakaryocytic blast crisis as a first presentation of chronic myeloid leukemia. Eur J Haematol. 2002;69:58- 61.
12. Wu CD, Medeiros LJ, Miranda RNy col. Chronic myeloid leukemia manifested during megakaryoblastic crisis. South Med J. 1996;89:422-7.
13. Campiotti L, Grandi AM, Biotti MG y col. Megakaryocytic blast crisis as first presentation of chronic myeloid leukemia. Am J Hematol. 2007;82:231-3.
14. Pullarkat ST, Vardiman JW, Slovak ML y col. Megakaryocytic blast crisis as a presenting manifestation of chronic myeloid leukemia. Leuk Res. 2008 Nov;32(11):1770-5. doi: 10.1016/j.leukres.2008.02.025.
15. Karkuzhali P, Shanthi V, Usha T. A case of chronic myeloid leukaemia presenting as megakaryocytic blast crisis (AML M7). Ecancermedicalscience. 2013 Nov 21;7:375. doi: 10.3332/ecancer.2013.375.
16. Khemka R, Gupta M, Jena NK. CML with Megakaryocytic Blast Crisis: Report of 3 Cases. Pathol Oncol Res. 2019 Jul;25(3):1253-1258. doi: 10.1007/s12253-018-0484-7.
17. Kashima E, Nakano E, Watanabe Y y col. Acute Megakaryoblastic Leukemia Harboring a Subclone Expressing BCR-ABL1 Fusion Gene Product. Intern Med. 2021 Nov 15;60(22):3609-3614.
18. Pemmaraju N, Kantarjian H, Sweet K y col. North American Blastic Plasmacytoid Dendritic Cell Neoplasm Consortium: position on standards of care and areas of need. Blood. 2023 Feb 9;141(6):567-578. doi: 10.1182/blood.2022017865.
19. Lee YJ, Kim Y, Park SH, Jo JC. Plasmacytoid dendritic cell neoplasms. Blood Res. 2023 Apr 30;58(S1):90-95. doi: 10.5045/br.2023.2023052.
20. Adams RL, McCarthy C, Bird RJ. Philadelphia chromosome-positive blastic plasmacytoid dendritic cell leukemia. Cancer Genet Cytogenet. 2009 Dec;195(2):186-8. doi: 10.1016/j.cancergencyto.2009.08.012.
21. Suzuki H, Takeshita M, Hirai R y col. Blastic Plasmacytoid Dendritic Cell Neoplasm Developed in Chronic Myeloid Leukemia in Molecular Remission During a Four-Year Treatment-Free Interval After Six Years of Dasatinib Treatment. Cureus. 2024 Jun 8;16(6):e61944. doi: 10.7759/cureus.61944.
22. Shah NP, Bhatia R, Altman JK y col. Chronic Myeloid Leukemia, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2024 Feb;22(1):43-69. doi: 10.6004/jnccn.2024.0007.
23. Brioli A, Lomaia E, Fabisch C y col. Management and outcome of patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era - analysis of the European LeukemiaNet Blast Phase Registry. Leukemia. 2024 May;38(5):1072-1080. doi: 10.1038/s41375-024-02204-y.
24. Copland M. Treatment of blast phase chronic myeloid leukaemia: A rare and challenging entity. Br J Haematol. 2022 Dec;199(5):665-678. doi: 10.1111/bjh.18370.