Enfoque de un paciente con perfil de hierro sugestivo de sobrecarga
Approach to a patient with an iron profile suggestive of overload
Chiappe G
Servicio Hematología, Hospital Milstein. Buenos Aires, Argentina.
ORCID Gustavo Chiappe: 0009-0007-3320-1342
Palabras claves: hiperferritinemia,
sobrecarga de hierro,
hemocromatosis.
Keywords: hiperferritinemia,
iron overload,
hemochromatosis.
Resumen
Con frecuencia un perfil básico de hierro orienta hacia condiciones ferropénicas o de secuestro inflamatorio del hierro, pero a veces también hacia la sospecha de patologías con sobrecarga de hierro, que pueden o no terminar confirmándose. Por lo tanto, es necesaria una interpretación precisa de los resultados para evitar errores diagnósticos. La ferremia refleja la cantidad de hierro en tránsito en un momento determinado desde células que expresan la ferroportina hacia las que expresan el receptor de transferrina-1, mientras que la transferrinemia refleja la avidez del organismo por el hierro. Pero la ferritinemia puede ser reflejo tanto de los depósitos de hierro como de condiciones inflamatorias, con la consiguiente dificultad para interpretar sus resultados normales o elevados. En este articulo diferencio, entre los perfiles de hierro sugestivos de sobrecarga, los que cursan con una sobrecarga de hierro evidente de los que presentan sólo una hiperferritinemia sin evidencia (franca) de sobrecarga de hierro, aunque la superposición entre ambas situaciones es frecuente. La hiperferritinemia secundaria reactiva es, por mucho, más frecuente que la vinculada a sobrecarga de hierro, así como es común encontrar pacientes con más de una causa de hiperferritinemia. "Hiperferritinemia reactiva de origen desconocido" puede ser un rótulo diagnóstico provisorio para aquellos pacientes sin una causa (hasta el momento) evidente de su hiperferritinemia. Algunos pacientes tienen claramente presencia o ausencia de sobrecarga de hierro, pero en muchos casos la situación es dudosa, incompleta o intermitente. En consecuencia, es a veces difícil decidir sobre la indicación o no de una terapia quelante. La sobrecarga de hierro, de estar presente, puede ser secundaria (básicamente a patologías con eritropoyesis inefectiva) o primaria. Ésta, a su vez, puede ser clasificada como hemocromatósica (HFE o no HFE) o no hemocromatósica. Aunque muy raras, las sobrecargas de hierro primarias no hemocromatósicas tienen perfiles de hierro confundentes, pero fácilmente interpretables si se las sospecha. Una de ellas, la enfermedad por ferroportina, en las antípodas de las hemocromatosis, merece ser tenida en cuenta, ya que no es muy infrecuente. Finalmente, el diagnóstico de una hemocromatosis debe estar apoyado en un perfil de hierro inequívoco y confirmado por mutaciones HFE severas bialélicas o, más raramente, por mutaciones en genes no HFE. Por el contrario, mutaciones HFE leves, como la H63D, tanto en condición mono como bialélica, no justifican por sí solas una sobrecarga de hierro típica. Frente a esta situación debemos investigar otra(s) causa(s) de sobrecarga de hierro: hemocromatosis no HFE si el perfil de hierro es típicamente hemocromatósico, o sobrecargas de hierro no hemocromatósicas (primarias o secundarias) en caso contrario. El riesgo de considerar una mutación HFE leve (aún en la condición doble heterocigota HFE C282Y/H63D) como responsable única de la sobrecarga de hierro o de una hiperferritinemia, es dejar de lado otras condiciones que pueden merecer atención y tratamiento quizás más prioritarios.
Abstract
A basic iron profile often provides a useful approach to ferropenic or iron-sequestered inflammatory conditions, but sometimes also to the primary suspicion of iron overloading pathologies, which may or may not, at last, harbor a real iron overload. Therefore, a thoughtful interpretation of the results is mandatory to avoid misdiagnosis. The serum iron concentration reflects the quantity of iron moving at one moment from ferroportin- to transferrin receptor-1-expressing cells, while the transferrin concentration reflects the organism avidity for iron. But serum ferritin concentration may reflect iron deposits as well as inflammatory conditions, hence being difficult the interpretation of its normal or high values. In this paper I distinguish, among abnormal iron profiles, those with evident iron overload from those with hiperferritinemia without (clear) evidence of iron overload, although there is a frequent overlap between these two conditions. Secondary reactive hiperferritinemia is, by far, much more common than hiperferritinemia related to iron overload, as it is also frequent to deal with patients with more than one cause for their hiperferritinemia. "Reactive hiperferritinemia of unknown origin" should be the temporary diagnosis for patients with (yet) no evident etiology for their hiperferritinemia. Some patients have clearly absence or presence of iron overload, but in many cases this condition is doubtful, incomplete or intermittent. Hence, it is difficult in some cases to decide the need for chelation therapy. Iron overload, if present, may be secondary (basically to ineffective erythropoiesis) or primary. Primary iron overload may be, in turn, classified as hemochromatosic (HFE or non HFE) or non hemochromatosic. Although very rare, the non hemochromatosic primary iron overloads have confounding iron profiles, but with an easy approach if suspected. One of them, the ferroportin disease, just the opposite of hemochromatosis, deserves some attention, for it is not very infrequent. Finally, the diagnosis of hemochromatosis must be supported by an unequivocal typical iron profile and by biallelic severe HFE mutations or, rarer, mutations in non-HFE genes. The last but not the least, mild HFE mutations, as H63D, either mono o biallelic, do not justify by themselves any typical iron overload. In presence of such a genetic diagnosis, other cause(s) of iron overload must be looked for: non-HFE hemochromatosis if the iron profile is typically hemochromatosic, non-hemochromatosic iron overload (primary o secondary) if not. The risk of considering a mild HFE mutation (even in the case of HFE C282Y/H63D double heterozygous) as responsible for an iron overload or a hiperferritinemia is to overlook other causes for these conditions that may deserve particular and, eventually, more imperative attention and treatment.
1. Introducción
Tan abundante es el hierro en la corteza terrestre y tan peligroso cuando está libre, que el genoma humano tiene unos 400 genes (o sea un 2% del genoma) que codifican para proteínas vinculadas con su metabolismo. Esta actitud "defensiva" incide en que haya una cantidad mucho mayor de personas con ferropenia (unos 2.000 millones en el mundo, la mitad en estadío clínico de anemia) que con sobrecarga de hierro. Ambas patologías son detectables a través de un perfil de hierro básico (ferremia, transferrinemia, ferritinemia), pero mientras un componente ferropénico o inflamatorio (como en la anemia de los procesos inflamatorios) son definibles a partir de un perfil típico, las sobrecargas de hierro son, frecuentemente, apenas sospechables a partir de un par de perfiles de hierro heterogéneos. De hecho, un porcentaje de saturación de la transferrina francamente elevado es altamente sugestivo de sobrecarga de hierro, pero una hiperferritinemia (incremento mucho más frecuente que el del porcentaje de saturación) puede acompañarse de cantidades de hierro en depósito normales, aumentadas o disminuidas. En la mayor parte de los pacientes la orientación es clara desde un principio, pero a veces es difícil decir con certeza si el paciente tiene o no una sobrecarga real de hierro, con las implicancias terapéuticas consecuentes.
Un poco arbitrariamente, la línea divisoria la pongo entre "hiperferritinemias sin sobrecarga (franca) de hierro" y "sobrecargas de hierro", los dos capítulos que desarrollo luego de una breve introducción sobre el perfil de hierro.
2. Perfil de hierro
El perfil de hierro básico incluye:
- la ferremia y la transferrinemia, reflejo del hierro plasmático y
- la ferritinemia, reflejo del hierro en depósito.
Además de ellos hay otros parámetros para evaluar el estado del hierro en el organismo: protoporfirina eritrocitaria libre o unida al zinc, receptor soluble de transferrina, hepcidinemia, evaluación de hemosiderina y sideroblastos mediante la reacción de Perls en un frotis de médula ósea, resonancia nuclear magnética, etc., pero todos ellos se sitúan en una segunda línea diagnóstica.
2.1. Ferremia y transferrinemia
El hierro plasmático (3 mg) representa menos de una milésima parte del hierro corporal total (3-4 g). Es el hierro que está en tránsito desde células que expresan la ferroportina en su membrana plasmática (enterocitos, macrófagos, hepatocitos, eritroblastos terminales, trofoblastos placentarios) hacia las células que expresan el receptor de transferrina-1 (todas las células del organismo).
Mientras que el egreso del hierro plasmático está regulado por las necesidades de hierro de cada célula, reflejadas en el grado de expresión en su superficie del receptor de transferrina-1, su ingreso al plasma va a depender de la presencia o no de hepcidina sérica que, al unirse a su receptor, la ferroportina, bloquea su capacidad exportadora de hierro, y luego ambas se internalizan para ser degradadas por ubiquitinación a nivel proteasómico.
A su vez, la síntesis de hepcidina en el hepatocito está regulada:
a) positivamente por
- el hierro sérico, vía receptor de transferrina-2,
- el hierro intra células estrelladas hepáticas, vía proteínas morfogénicas óseas-6 y -2,
- el componente inflamatorio, vía interleuquina 6, y
b) negativamente por
- el hierro intra hepatocito, vía matriptasa y
- la eritropoyesis inefectiva, vía eritroferrona.
Para atravesar membranas o para integrarse a la protoporfirina IX para constituir el hemo, el hierro debe estar en estado ferroso (Fe++), mientras que para su transporte (transferrina) o almacenamiento intracelular (ferritina) debe estar en estado férrico (Fe+++). Su oxidación al momento de ingresar al plasma es llevada a cabo por la hefestina (en el borde laterobasal del enterocito) o por la ceruloplasmina plasmática (o unida vía puente glicolipídico a la membrana celular en el tejido nervioso), mientras que su reducción al egreso del plasma es llevada a cabo, intra endosomas invaginados, por la proteína Steap-3.
La transferrina es la proteína plasmática encargada de que el viaje del hierro en el plasma se haga en condiciones de extrema seguridad. La transferrina tiene una gran avidez por el hierro, y es capaz de transportar hasta dos átomos de hierro por molécula. O sea que en el plasma la transferrina puede estar en forma de:
- apotransferrina (50%): transferrina vacía de hierro,
- transferrina monoférrica (40%): transferrina que transporta sólo un átomo de hierro, o
- transferrina diférrica (10%): transferrina que transporta dos átomos de hierro.
O sea que 10 moléculas de transferrina, que podrían llegar a transportar hasta 20 átomos de hierro, en la práctica sólo transportan 6. Por eso decimos que el porcentaje de saturación normal de la transferrina es del 30%. El 70% de capacidad de transporte de hierro latente es un amplio margen de seguridad para que cualquier átomo de hierro libre en plasma, terriblemente tóxico, sea inmediatamente captado por la transferrina. En condiciones de sobrecarga de hierro, con saturaciones ya por encima del 80%, es posible detectar la presencia de cierta cantidad de hierro libre en plasma (non transferrin bound iron).
Como vimos en el ejemplo anterior, el porcentaje de saturación de la transferrina se calcula dividiendo la cantidad de hierro que la transferrina podría transportar si estuviese saturada al 100% (lo que se denomina su capacidad de transporte) por la cantidad que realmente transporta (ferremia).
Sabemos cómo se mide la ferremia, pero la capacidad de transporte puede medirse de dos maneras distintas:
- clásicamente, saturando la muestra con hierro, eliminando el excedente y dosando una nueva ferremia, y
- actualmente, en forma indirecta, dosando la concentración sérica de la transferrina y calculando, con una elemental regla de tres simple, la capacidad de transporte, sabiendo que 1 mg de transferrina sérica es capaz de transportar, con una saturación del 100%, 1.27 µg de hierro.
El problema es que, no infrecuentemente, los laboratorios informan el resultado de la "transferrinemia" como "capacidad de transporte" (o viceversa) o, peor aún, calculan el porcentaje de saturación de la transferrina dividiendo el valor de la ferremia por el de la transferrinemia, algo totalmente incorrecto. Si tenemos duda con respecto a la rotulación de los resultados informados, guiémonos por las unidades: si están en mg/dL seguramente el resultado numérico corresponde a la transferrinemia, y en este caso debemos hacer nosotros la regla de tres simple (en la práctica, multiplicar por 1.27 y acomodar la coma) para obtener la capacidad de transporte, que nos va a servir de denominador para calcular el porcentaje de saturación de la transferrina, con la ferremia como numerador.
La ferremia refleja la cantidad de hierro que hay en un momento determinado en el plasma, en tránsito desde su lugar de origen hacia su lugar de destino. La transferrinemia, en cambio, refleja la avidez del organismo por el hierro: alta en caso de ferropenia, baja cuando no la hay.
La ferremia, además de su ritmo circadiano, tiene diversos factores moduladores, así como eventuales errores de procedimiento. O sea que es un dato débil, mientras que la transferrinemia, especialmente si es medida directamente (y no a partir de la capacidad de transporte), es un dato fuerte y de interpretación fidedigna. Con frecuencia, frente a perfiles séricos de hierro dudosos, es preferible guiarse por la transferrinemia antes que por la ferremia.
Además, recordando que el porcentaje de saturación no es más que un cociente, siempre es de utilidad diferenciar si un cociente anormal se debe más a una alteración del numerador (ferremia) o del denominador (capacidad de transporte).
2.2. Ferritinemia
Una ferritinemia baja es, salvo en presencia de una carencia severa de ácido ascórbico, prácticamente sinónimo de ferropenia. La interpretación de una ferritinemia normal o alta puede ser, por el contrario, dificultosa, dada la variedad de causas que la pueden deparar.
2.2.1. Ferritina. Conceptos fisiológicos
Tan popular en la clínica médica como enigmática en sus orígenes, mecanismos regulatorios de síntesis y funciones (alguna vez me pregunté si no es un ñoqui, sin trabajo específico asignado), la ferritina sérica sigue envuelta en una nube de dudas y controversias.
La ferritina es una proteína eminentemente intracitoplasmática, de estructura globular compuesta por 24 cadenas de tipo H (pesadas: 183 aminoácidos, 21 kDa) y L (livianas: 175 aminoácidos, 19.5 kDa), en 25 combinaciones posibles (0-24 cadenas H, el resto L), con predominio de cadenas H en las ferritinas ácidas, abundantes en tejidos de alto requerimiento metabólico (corazón - Hearth), y de L en las básicas, abundantes en tejidos con mayor función de depósito (hígado - Liver).
Este caparazón proteico puede almacenar en su interior hasta unos 3.000-4.500 átomos de hierro en forma de hidroxifosfato férrico. El ingreso del hierro está manejado por las cadenas H, las únicas con capacidad de oxidar el hierro de ferroso a férrico (que es como se va a almacenar), mientras que las cadenas L participan en la mineralización del hierro ingresado. El egreso del hierro de la ferritina parece ser exclusivamente vía ferritinofagia en autofagolisosomas, cadalso al que la ferritina es llevada por la proteína Ncoa4 (Nuclear receptor coactivator 4). O sea que el camino de ingreso del hierro (a una molécula como la ferritina, a cualquier célula o al organismo en su conjunto) está frecuentemente más "alfombrado" que el de egreso.
Pero además de trabajar intracelularmente como reservorio de hierro, la ferritina sérica tiene otra ocupación en sus horas libres. Como reactante de fase aguda positivo, integra ese grupo de proteínas séricas vinculadas, a la vez, con el metabolismo del hierro y con la lucha antiinfecciosa: la hepcidina (descrita originalmente como una proteína antibacteriana de origen hepático, de ahí su nombre), la transferrina (reactante de fase aguda negativo) y la lactoferrina, entre otras.
De hecho, la síntesis de la ferritina está regulada por condiciones tanto inflamatorias como vinculadas con el metabolismo del hierro:
- a nivel transcripcional por citoquinas inflamatorias: factor de necrosis tumoral-α, interleuquinas 1b y 6, vía activación del factor nuclear kB(1) y
- a nivel post transcripcional por las proteínas reguladoras del hierro (IRP1 y 2: iron-responsive proteins), que bloquean la traducción de su ARNm a nivel ribosómico uniéndose al elemento IRE (IRP-responsive element) que tienen en su región promotora a 5' del gen. Este mecanismo regulador parece ser válido a nivel macrofágico, pero no a nivel del hepatocito o de las células del epitelio intestinal(2).
La ferritina sérica, aparente hija descarriada de la ferritina intracitoplasmática, parecería estar vagando por el plasma sin saber bien qué hacer ni para dónde ir. Son más las dudas que las certezas.
Parece certero que la ferritina sérica está compuesta casi exclusivamente por cadenas L (las H serían sólo una minoría), que las cadenas L están glicosiladas para garantizarle a la ferritina una vida media más prolongada (tienen un sitio de N-glicosilación cerca de su extremo amino, responsable del aumento de su peso molecular de 19.5 a 23 kDa), y que prácticamente está vacía de hierro. Sus cadenas están codificadas por los mismos genes (FTL y FTH) que la ferritina citoplasmática, de la que parece ser su versión "externalizada". Y aquí comienzan las dudas.
¿Cómo llega la ferritina intracitoplasmática al plasma? No parece haber un mecanismo de secreción convencional, vía retículo endotelio, ya que ni las cadenas L ni las H tienen un péptido señal. Pese a ello sí parecería haber un pasaje por el aparato de Golgi para ser glicosilada, aunque Golgi y glicosilación no son estrictamente sinónimos (la albúmina pasa por el Golgi y en plasma no está glicosilada) y en ratones la ferritina sérica parece no estar glicosilada(2).
Sí parecería haber un mecanismo de secreción no convencional (unconventional protein secretion) vía vesículas extracelulares o, particularmente, lisosomas. Una hipótesis, a partir de modelos murinos, postula que un contingente especializado de la ferritina citoplasmática es destinado a una actividad protectora del hierro reactivo en los lisosomas. Este contingente ferritínico, con cadenas tanto H como L y conteniendo hierro, termina siendo liberado al plasma por vía lisosómica y rápidamente recaptado por células que expresan receptores de cadena H (como parece ser el caso del receptor de transferrina 1, con especificidad por cadenas H, o la proteína Scara5, con mayor preferencia por las cadenas L que H)(3), quedando en el plasma sólo la ferritina pobre en cadenas H, o sea rica en cadenas L y, ¿por ende?, pobre en hierro(2).
Por el contrario, la ferritina sérica no glicosilada posiblemente provenga de la lisis celular y sea realmente una proteína intracitoplasmática vertida "patológicamente" hacia el plasma.
¿Desde qué células es externalizada fisiológicamente la ferritina sérica? Clásicamente se postula que desde macrófagos y hepatocitos, pero en el mismo modelo murino mencionado antes, la ferritina sérica parece sólo provenir de macrófagos y de células del epitelio tubular renal (¿reabsorción?), no de hepatocitos(2).
¿Qué función cumple la ferritina en el plasma? Parece más vinculada con procesos antiinflamatorios (por ejemplo promover la netosis a partir de la unión a Msr1, su receptor específico en neutrófilos), inmunológicos, de señalización, de angiogénesis, etc.(3) que con el metabolismo del hierro (¿secuestro de hierro libre no unido a transferrina, función redundante con la de la apotransferrina?)(1).
Tal vez guarde relación con el metabolismo del hierro allende la barrera hemato-encefálica, como sugiere la acumulación cerebral patológica de hierro, especialmente en ganglios basales -pero con hipoferritinemia de este lado de la barrera-, con clínica extrapiramidal, en familias con neuroferritinopatías por mutaciones con alteración del encuadre en el exón 4 -último- del gen FTL(4-6) (Tabla 2).
2.2.2. Hiperferritinemia. Mecanismos fisiopatológicos
Se repite con frecuencia que, en ausencia de patologías inflamatorias, la ferritina sérica es reflejo fiel del hierro en depósito: cada µg/L de ferritina sérica correlaciona con 8 mg de hierro en depósito. Lo que no queda del todo claro es si la correlación es con el hierro total en el organismo o específicamente con el hierro en depósito intramacrofágico(2). Así parece sugerirlo la discrepancia entre los niveles de ferritina en las talasemias transfusión y no transfusión dependientes, con acumulación del hierro predominantemente en macrófagos y hepatocitos respectivamente. La correlación "imperfecta" en las talasemias no transfusión dependientes sugiere que la ferritinemia refleja más fielmente los depósitos macrofágicos que los parenquimatosos.
Además de incrementar su concentración sérica en respuesta a mecanismos "fisiológicos", ocasionalmente la ferritina también puede aumentar en plasma en condiciones "patológicas" de citólisis, por simple "vaciamiento" del contenido citoplasmático. En este caso la ferritina tiene proporciones variables de cadenas H y L y, al no haber pasado evidentemente por el aparato de Golgi, no está glicosilada, con una vida media consecuente más corta. La circunstancia de que sólo la ferritina glicosilada se une a la lectina concavalina-A permite su dosaje diferencial. Normalmente un 50-80% de la ferritina sérica está glicosilada, pero en la enfermedad de Still, por ejemplo, su porcentaje es menor del 20%(7).
3. Perfiles de hierro anormales
Distintas alteraciones en un perfil de hierro básico pueden orientarnos hacia 4 panoramas diferentes:
a) evidencia de componente ferropénico: ferremia baja, transferrinemia elevada, porcentaje de saturación francamente disminuido y ferritinemia baja.
b) evidencia de componente inflamatorio: ferremia baja (por hiperhepcidinemia), transferrinemia baja, porcentaje de saturación más o menos normal, ferritinemia normal o elevada.
c) sugerencia de sobrecarga de hierro: ferremia aumentada, transferrinemia algo baja, porcentaje de saturación francamente (> 50-60%) aumentado, ferritinemia en general aumentada. Léase: el aumento del porcentaje de saturación predomina por sobre el de la ferritinemia.
d) sugerencia de hiperferritinemia secundaria (reactiva) o primaria (hereditaria): ferritinemia aumentada, con porcentaje de saturación de la transferrina normal o apenas aumentado (digamos entre 40 y 60%, habitualmente oscilante, ya sea a lo largo del tiempo en un mismo paciente o en distintos pacientes con igual patología). Léase: el aumento de la ferritinemia predomina por sobre el del porcentaje de saturación.
Por eso, un poco arbitrariamente, al panorama d) lo voy a mencionar como "hiperferritinemia sin sobrecarga (franca) de hierro" (Sección 3.1, Tabla 1), mientras que al c) lo voy a rotular como "sobrecarga de hierro" (Sección 3.2, Tabla 3), dado que aquí la elevación del porcentaje de saturación es franca.
Un porcentaje de saturación de la transferrina francamente elevado es sinónimo de sobrecarga de hierro, pero la inversa no lo es: hay sobrecargas de hierro con porcentajes no elevados. Con la ferritinemia, en cambio, todas las variantes son posibles, desde hiperferritinemias severas sin la menor sobrecarga de hierro, hasta sobrecargas de hierro con ferritinemias normales. Las mayores "discrepancias" entre perfil de hierro y sobrecarga real las vamos a encontrar sobre todo en el capítulo de las hiperferritinemias reactivas, pero también en el de las sobrecargas de hierro no hemocromatósicas.
Más que el "jardín de los senderos que se bifurcan" de los algoritmos, es la conjunción de una serie de enfoques diagnósticos lo que más ayuda en la orientación diagnóstica: clínica (siempre soberana), perfil de hierro, presencia o no de anemia, el patrón de herencia, etc.
De presentar el paciente anemia, hay que tener en cuenta que:
- nunca deparan anemia ni las hemocromatosis ni las hiperferritinemias aisladas por mutaciones en el gen FTL.
- siempre está presente, y más bien severa -habitualmente por eritropoyesis inefectiva-, en las sobrecargas de hierro secundarias.
- también está presente -anemia hierro restricta- en las sobrecargas de hierro primarias no hemocromatósicas (en forma latente en la enfermedad por ferroportina, instalándose sólo al iniciar tratamiento quelante con sangrías demasiado frecuentes).
- su presencia en las hiperferritinemias reactivas dependerá en cada caso de la patología de base, con anemia de los procesos inflamatorios como la más característica.
Y respecto al patrón de herencia, no hay que olvidar que varias de las sobrecargas de hierro genéticamente determinadas (mutaciones en SLC40A1, FTL y FTH), aunque raras, son de herencia autosómica dominante, así que nunca hay que dejar de armar un buen árbol genealógico.
3.1. Hiperferritinemia sin sobrecarga (franca) de hierro
El tema de los diagnósticos diferenciales en pacientes con hiperferritinemia ha sido revisado recientemente por diversos autores(8-16). Comprende dos grandes capítulos (Tabla 1):
- causas genéticas (Sección 3.1.1. "Hiperferritinemias aisladas") y
- causas adquiridas (Sección 3.1.2. "Hiperferritinemias reactivas").
Tabla 1. Causas de hiperferritinemia sin sobrecarga (franca) de hierro
|
Tabla 1. Causas de hiperferritinemia sin sobrecarga (franca) de hierro |
|
|
Genéticas |
Por aumento de síntesis, con un 100% de apoferritina glicosilada (ferritina L) = hiperferritinemia aislada: |
|
- síndrome hiperferritinemia/cataratas congénitas - hiperferritinemia "benigna" |
|
|
Adquiridas "reactivas" |
Por aumento de la síntesis/secreción, con predominio de apoferritina glicosilada (ferritina L): |
|
- causas "inflamatorias" - síndrome metabólico, DIOS (dysmetabolic iron overload syndrome) - etilismo - neoplasias: histiocitosis maligna, carcinomas de pulmón, mama, ovario, riñón, linfoma, liposarcoma - enfermedad de Gaucher - histiocitosis reactiva - (porfiria cutánea tarda) |
|
|
Por aumento de la liberación desde células dañadas (citólisis), con presencia tanto de ferritina glicosilada como no glicosilada: |
|
|
- esteatosis hepática y esteatohepatitis - hepatitis viral crónica - cirrosis hepática - necrosis hepática masiva por sepsis, hepatitis aguda o daño tóxico - patologías autoinmunes - infecciones agudas o crónicas - infarto agudo de miocardio - infarto esplénico |
|
|
La subclasificación de las patologías "reactivas" según su mecanismo fisiopatológico más probable (aumento de síntesis/secreción vs. citólisis) es un poco arbitrario y de ninguna manera estricto. En cursiva figuran algunas patologías que, con más frecuencia, suelen, aunque no siempre, acompañarse de una sobrecarga leve/moderada de hierro. La porfiria cutánea tarda figura entre paréntesis porque su relación con el hierro, su factor predisponente más importante, es sólo unidireccional. |
|
3.1.1. Hiperferritinemia aislada (congénita)
Engloba 2 patologías: - el síndrome hiperferritinemia hereditaria con cataratas, y
- la hiperferritinemia benigna, mucho menos frecuente que la anterior.
El gen FTL es prolijo en cuanto a sus mutaciones y prolífico en cuanto a la variedad de cuadros clínicos que ellas deparan(17):
Tabla 2. Defectos en el gen FTL
|
Tabla 2. Defectos en el gen FTL |
|||
|
Patología |
Herencia |
Tipo de mutación |
Ferritinemia |
|
síndrome hiperferritinemia cataratas |
dominante |
en IRE a 5' |
elevada |
|
hiperferritinemia benigna |
dominante |
con sentido equivocado en exón 1 |
elevada |
|
neuroferritinopatía |
dominante |
con alteración del encuadre en exón 4 (último) |
< 12 µg/L |
|
deficiencia de ferritina |
dominante |
en el codón de inicio o que afecta el empalme, |
< 12 µg/L |
|
deficiencia de ferritina |
recesiva |
sin sentido |
< 1 µg/L |
Fisiológicamente, en caso de ferropenia una cantidad mayor de IRPs 1 y 2 se unen al IRE (único) presente a 5' de los ARNm de las cadenas de ferritina, bloqueando su traducción a nivel ribosómico. En consecuencia, mutaciones en el IRE a 5' de los genes de ferritina van a impedir la correcta unión de las IRPs, deparando una hiperproducción descontrolada de cadenas de ferritina. Pero mientras que la hiperproducción de cadenas livianas (mutaciones en el IRE de FTL) son inocentes (síndrome de hiperferritinemia cataratas), la hiperproducción de cadenas pesadas (mutaciones en el IRE de FTH, un solo caso descrito) sí cursa con sobrecarga de hierro, algo lógico teniendo en cuenta que las cadenas H son las que tienen actividad ferroxidante, imprescindible para que el hierro pueda ingresar al caparazón ferroportínico.
En el síndrome hiperferritinemia cataratas el exceso de ferritina plasmática termina depositándose en el cristalino, determinando, como única manifestación clínica, cataratas bilaterales en edades tempranas y con características oftalmológicas específicas, pero sin la menor sobrecarga de hierro.
Una hiperferritinemia aislada franca acompañada de estas cataratas precoces en el marco de un patrón de herencia dominante es altamente sugestivo y prácticamente diagnóstico del síndrome hiperferritinemia cataratas, sin necesidad imprescindible de recurrir al estudio genético para su confirmación. Su única conducta terapéutica es la resolución quirúrgica de las cataratas.
En ausencia (confirmada por oftalmólogo) de cataratas, puede tratarse de una hiperferritinemia benigna, mucho menos frecuente, por mutaciones con sentido equivocado en el exón 1 (T30I) del gen FTL(18). Aquí sí, el estudio genético es necesario para su confirmación, pero tampoco depara patología clínica ni requiere tratamiento.
Por el contrario, las neuroferritinopatías(15) cursan con niveles bajos de ferritina en sangre, pero con depósitos de ferritina y de hierro del otro lado de la barrera hematoencefálica.
3.1.2. Hiperferritinemias "reactivas"
Concepto fundamental: la hiperferritinemia "reactiva" es mucho más frecuente que la vinculada con una sobrecarga de hierro, su espectro diagnóstico mucho más amplio y complejo y su ámbito más clínico que hematológico. Como hematólogos no nos podemos permitir no sospechar una posible porfiria cutánea tarda o un Gaucher, pero lo más probable es que el paciente sea portador de una hepatopatía (etílica, viral, etc.), de un síndrome metabólico (sin o con cierto grado leve de sobrecarga de hierro: DIOS -dysmetabolic iron overload syndrome-), síndrome de ovario poliquístico, o de un listado amplio de patologías inflamatorias, infecciosas o autoinmunes. El resto del cuadro clínico nos va a orientar en la estrategia a seguir en cada caso. En la frondosidad de este bosque de diagnósticos diferenciales hay que tratar de no perder de vista una correlación lógica entre la severidad de la(s) patología(s) de base y los niveles de la hiperferritinemia.
Más del 40% de los pacientes con hiperferritinemia tienen más de una causa que la justifique(7), todas adquiridas o, eventualmente, con la colaboración de algún defecto hereditario sin relevancia clínica per se, pero que puede aportar algún granito de arena a los niveles de ferritinemia: típicamente patología clínica "reactiva" acompañada de la presencia mono o bialélica de la mutación HFE leve (H63D), suficiente, quizás, para agravar algo el perfil férrico, pero de ninguna manera para deparar un patrón hemocromatósico.
Es frecuente que, después de un estudio más o menos exhaustivo, no se encuentre patología clínica evidente que justifique la hiperferritinemia. Entonces uno puede enfocar hacia otras causas de hiperferritinemia sin evidencia (franca) de sobrecarga de hierro, o hacia sobrecargas con perfiles de hierro complejos, como las no hemocromatósicas. Pero de no tener éxito tampoco por estos territorios, hay que recordar que el de las hiperferritinemias reactivas es un capítulo que nunca se cierra, ya sea porque no se pudo/supo diagnosticar correctamente una patología responsable, o porque ésta, "silente" de entrada, evidente sólo a través de sus reactantes de fase aguda (ferritinemia), recién más tardíamente aflora con el resto de su cuadro clínico (un cáncer oculto como patología más típica, o una enfermedad reumatológica de instalación insidiosa). Por eso, descartados, hasta donde se pueda, otros diagnósticos, el paciente puede quedar con el rótulo, siempre provisorio, de "hiperferritinemia reactiva de etiología -aún- no identificada", con la tranquilidad de la no necesidad, al menos inmediata, de un tratamiento quelante, pero fusil en mano a la espera de ver por dónde puede saltar la liebre.
3.1.2.1. Hepatopatías
Sin lugar a dudas, el hepatocito es la célula más compleja del organismo. Hepatopatías crónicas (hígado graso, hepatopatía etílica, viral, cirrosis, etc.) suelen cursar con alteración de los parámetros del hierro, frecuentemente con cierto grado de sobrecarga, y gran dificultad para definir qué es causa y qué consecuencia. Seguramente algo de círculo vicioso hay en esta relación entre hepatocito y hierro y, obviamente, toda hepatopatía amerita un estudio del perfil del hierro, así como toda alteración en el perfil de hierro amerita, al menos, un hepatograma.
3.1.2.2. Síndrome metabólico
Lo habitual en estos pacientes con una combinación de hipertensión, diabetes, hipertrigliceridemia, obesidad, esteatohepatitis y/o hígado graso, es que presenten una hiperferritinemia acompañada de un porcentaje de saturación de la transferrina normal (o ligeramente aumentado, en general más por disminución del denominador que por aumento del numerador). En algunos pacientes puede estar presente una sobrecarga real de hierro (DIOS: dysmetabolic iron overload syndrome) que, obviamente, tiene que ser confirmada fehacientemente. Lamentablemente, en muchos trabajos se rotula a estos pacientes como "portadores de una sobrecarga de hierro", con la consecuente indicación quelante, por el sólo hecho de presentar una ferritinemia elevada. Recomendación salomónica frecuente en estos pacientes con perfil de hierro dudoso: proponerles ser dadores altruistas de sangre.
3.1.2.3. Porfirias
Hierro es mala palabra en las porfirias erosivas crónicas, particularmente la cutánea tarda (PCT). No es que ésta predisponga a la sobrecarga de hierro, sino que el hierro es, por mucho, el factor adquirido más importante, con presencia o no de un defecto en el gen UROD, en la fisiopatología de sus formas hereditaria y esporádica, respectivamente. Por este motivo las mutaciones hemocromatósicas son más frecuentes en pacientes con PCT que en la población general: 20% de los pacientes con PCT son homocigotas para la mutación HFE C282Y y 7% doble heterocigotas HFE C282Y y H63D.
En presencia de un exceso de hierro, el citocromo p450 convierte al uroporifirnógeno en uroporfometeno, que la uroporfirinógeno decarboxilasa adopta como sustrato, pero, sin poder decarboxilarlo, por lo que termina bloqueada e inhibida. Prácticamente no hay porfiria cutánea tarda sin cierto grado de sobrecarga de hierro, y las sangrías, originalmente indicadas como tratamiento sintomático para disminuir los niveles de porfirinas séricas, demostraron ser patogénicamente efectivas al disminuir también los niveles de hierro.
3.2. Sobrecarga de hierro
Dado que la cantidad de hierro en el organismo se regula sólo por ingreso, el común denominador de todas las sobrecargas de hierro es un ingreso excesivo de hierro por vía enteral o, eventualmente, parenteral (transfusiones).
Las sobrecargas de hierro las clasificamos en primarias y secundarias (Tabla 3). Las sobrecargas primarias son debidas a mutaciones en genes de proteínas ferrohemostáticas, o sea vinculadas con el metabolismo y la regulación del hierro. Las sobrecargas secundarias lo son a diferentes patologías, hereditarias o no hereditarias, que determinan una alteración funcional de las proteínas ferrohemostáticas.
Tabla 3. Causas de sobrecarga de hierro
|
Tabla 3. Causas de sobrecarga de hierro |
||||||
|
Primarias: por mutaciones en genes de proteínas ferrohomeostáticas |
||||||
|
Hemocromatosis |
Del adulto |
HFE |
Recesiva. Asociada generalmente a cofactores adquiridos |
|||
|
TFR2 |
Recesiva |
|||||
|
SLC40A1 |
Dominante (mutaciones con ganancia de función) Hepcidina aumentada |
|||||
|
Juvenil |
HJV |
Recesiva (2ª a mutaciones en PIGA(19)) |
||||
|
HAMP |
Recesiva |
|||||
|
Enfermedad por ferroportina |
SLC40A1 |
Dominante (mutaciones con pérdida de función) |
||||
|
Aceruloplasminemia |
CP |
Recesiva (2ª a mutaciones en PIGA(19)) |
||||
|
A / hipotransferrinemia |
TF |
Recesiva |
||||
|
Deficiencia de Steap3 |
STEAP3 |
Recesiva |
||||
|
Deficiencia de DMT1 |
DMT1 |
Recesiva |
||||
|
Mutación IRE a 5' FTH |
FTH |
Dominante(20) |
||||
|
Secundarias a patologías que inducen una expresión alterada de las proteínas ferrohomeostáticas |
||||||
|
Sistémicas |
Aporte enteral/parenteral excesivo. Ej: talasemias transfusión dependientes |
|||||
|
Eritropoyesis inefectiva |
Talasemias no transfusión dependientes |
|||||
|
Anemias diseritropoyéticas congénitas |
||||||
|
Anemias sideroblásticas hereditarias (+ sobrecarga focalizada mitocondrial) |
||||||
|
Otras eritropatías hereditarias: deficiencia PK, xerocitosis hereditaria, etc. |
||||||
|
Hepatopatía gestacional aloinmune ("hemocromatosis neonatal aloinmune") |
||||||
|
Focalizadas |
Anemias sideroblásticas hereditarias |
mitocondrias (+ sobrecarga sistémica) |
||||
|
Ataxia de Friedreich |
mitocondrias |
|||||
|
Hepatopatías crónicas |
parénquima, mesénquima (DIOS: dysmetabolic iron overload syndrome) |
|||||
|
Síndrome metabólico |
sistema retículo endotelial |
|||||
|
(Anemia) procesos inflamatorios |
sistema retículo endotelial |
|||||
|
Neuroferritinopatías |
citosol (ganglios basales) |
|||||
|
Enf. neurodegenerativas adquiridas |
ganglios basales (Alzheimer, Parkinson, etc.) |
|||||
|
Hemosiderosis pulmonar primaria |
pulmón |
|||||
Tal como vemos en la tabla 3, dejando de lado las sobrecargas secundarias focalizadas, en todas las demás hay una sobrecarga sistémica generalizada, sospechable/detectable a través del perfil de hierro, eventualmente con el apoyo de la evaluación de la concentración hepática de hierro. Típicamente en las sobrecargas primarias hemocromatósicas y frecuentemente en las secundarias sistémicas, el porcentaje de saturación de la transferrina elevado es el dato orientador más importante, pero en las sobrecargas primarias no hemocromatósicas el perfil de hierro suele ser más heterogéneo y prestar a confusión. En esa puja inflacionaria entre porcentaje de saturación de la transferrina y ferritinemia, la elevación del primero predomina en las sobrecargas primarias hemocromatósicas, mientras que la segunda es más universal en las sobrecargas primarias no hemocromatósicas. En principio podemos decir que el patrón oro de una sobrecarga de hierro sistémica es la concentración hepática de hierro (LIC: liver iron concentration), aunque rara vez sea necesario llegar a ella para confirmar su presencia.
3.2.1. Sobrecarga de hierro primaria
Las sobrecargas primarias de hierro pueden ser hemocromatósicas o no hemocromatósicas. En las hemocromatósicas el defecto radica en genes vinculados con la regulación de la absorción intestinal de hierro. En las no hemocromatósicas, en genes vinculados con el ingreso, transporte y egreso del hierro plasmático y con su almacenamiento.
3.2.1.1. Sobrecarga de hierro primaria hemocromatósica
Las hemocromatosis representan:
a) un síndrome clínico hereditario causado por los efectos tóxicos del hierro en órganos parenquimatosos: es una entidad bioquímica -estadío 1- o clínica -estadío 2-. El defecto genético por sí solo -estadío 0-, particularmente en las mutaciones HFE, NO define hemocromatosis, sólo predisposición.
b) por falla en la prevención del ingreso no deseado de hierro al plasma: tanto la incorporación de hierro al organismo como al plasma se regulan por ingreso, no por egreso.
c) en ausencia de requerimientos eritropoyéticos aumentados: descarta formas secundarias de sobrecarga de hierro que también cursan con hipohepcidinemia.
Actualmente se propone eliminar el adjetivo "hereditaria" de su denominación porque no existe la hemocromatosis adquirida(21): la patología clínica adquirida que más se le asemeja es la hepatopatía aloinmune gestacional (antes mal llamada "hemocromatosis neonatal"), en que la hipohepcidinemia se acompaña, entre otros defectos, de hipotransferrinemia, con un cuadro clínico muy complejo y severo.
La característica definitoria de las hemocromatosis es una actividad descontrolada de la ferroportina (habitualmente por déficit de hepcidina, pero también puede ser por mutaciones con ganancia de función en el gen SLC40A1, que codifica para la ferroportina). Esta actividad descontrolada de la ferroportina depara un vaciamiento del hierro desde las células que la expresan en su membrana (macrófagos, enterocitos, etc.), hacia el plasma. El de los macrófagos es hierro reciclado, pero el de los enterocitos es hierro captado desde la luz intestinal, normalmente a la espera del control de "migraciones" en la membrana basolateral, a cargo de la hepcidina, que es la que autoriza o no su ingreso real al organismo. Según el grado de hiperactividad de la ferroportina el ingreso de hierro "excedente" (por encima de los requerimientos basales) va a ser leve (mutaciones HFE, hemocromatosis del adulto típica), intermedio (mutaciones TFR2 o SLC40A1) o alto (mutaciones HJV o HAMP, hemocromatosis juvenil). Y este "excedente" diario de hierro comienza a ingresar al organismo desde el momento mismo de la activación funcional del intestino (nacimiento), ya que, durante el embarazo, al menos en sus primeras etapas, el destino prioritario del hierro materno es la placenta y no el feto.
En la hemocromatosis HFE (por mutaciones en el gen HFE), una de las variantes más leves de hemocromatosis, este "excedente" diario de hierro que se va incorporando al organismo no modifica ningún parámetro del perfil de hierro (estadío 0) durante años o décadas (Figura 1), hasta que comienza a ser evidente a través de un aumento de la ferremia y, en consecuencia, del porcentaje de saturación de la transferrina (estadío 1). Dado que en las hemocromatosis hay un vaciamiento compulsivo del hierro macrofágico, precisamente el que, en principio, más correlaciona con la ferritina sérica, es lógico, entonces, que el incremento de la ferritinemia sea más tardío que el inicio del incremento de la ferremia y del porcentaje de saturación: la ferritinemia recién comienza a elevarse cuando el porcentaje de saturación de transferrina supera el 70-80%. Tan es así, que la situación inversa, elevación de la ferritinemia sin elevación franca de la ferremia ni del porcentaje de saturación de la transferrina, es absurda en el marco de un cuadro hemocromatósico.
Figura 1. Hemocromatosis. Perfil de hierro
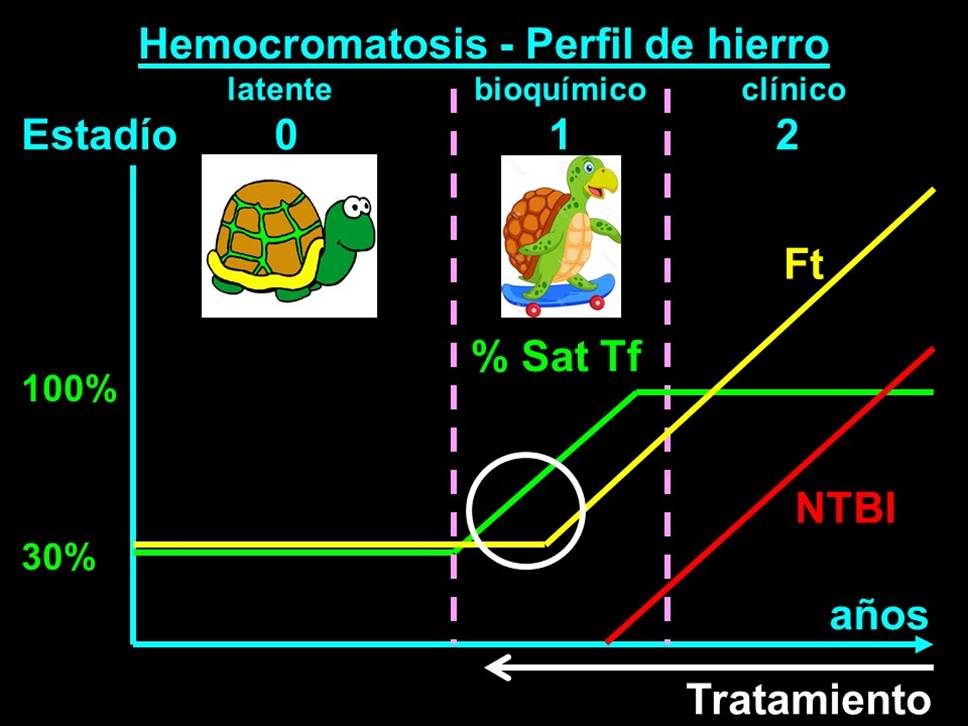
Figura 1. Hemocromatosis estadío 0 es cuando sólo está presente el defecto genético y la acumulación de hierro aún no es suficiente para deparar ninguna alteración en el perfil de hierro, mucho menos clínica. Dado que en la hemocromatosis HFE esta acumulación es muy lenta y habitualmente requiere de la concurrencia de otros factores adquiridos para alcanzar niveles de notoriedad, algo que sólo ocurre en una fracción (10-50% según los autores) de los individuos portadores que nacen con el defecto genético, tal vez no sea conveniente rotular de "hemocromatósicos" a estos pacientes, sino sólo de portadores del defecto genético que predispone a dicha condición. Hemocromatosis estadío 1 es cuando el perfil de hierro ya está alterado, pero aún sin consecuencias clínicas, que definen al estadío 2. Y el círculo blanco en el estadío 1 resalta cómo el aumento en el porcentaje de saturación de la transferrina antecede al comienzo del aumento de la ferritina. Y dado que en la hemocromatosis HFE este estadío 1 suele tardar años en alcanzar, eventualmente, el estadío 2, éste es el momento de detectarla mediante una medición cada tanto (¿unos 3-5 años?) del porcentaje de saturación de la transferrina que, de paso, nos va a destapar una cantidad muchísimo mayor de pacientes con ferropenia (latente o clínica).
Es muy didáctico comparar los cuadros clínicos y bioquímicos de las dos entidades que integran las sobrecargas de hierro ferroportínicas:
- hemocromatosis síndrome hemocromatósico por hiperactividad de la ferroportina,
- enfermedad por ferroportina síndrome ferroportínico por hipoactividad de la ferroportina.
Cara y cruz de una misma moneda, ambos defectos genéticos, funcionalmente opuestos, terminan determinando, por mecanismos fisiopatológicos bien diferentes, sobrecargas sistémicas de hierro.
La tabla 4 contrasta las características fisiopatológicas y clínicas de ambas.
Tabla 4. Síndromes hemocromatósico y ferroportínico: fisiopatológica y clínicamente opuestos
|
Tabla 4. Síndromes ferroportínico y hemocromatósico: fisiopatológica y clínicamente opuestos |
|||
|
Síndrome hemocromatósico |
Síndrome ferroportínico |
||
|
Actividad aumentada de ferroportina por hipohepcidinemia o por mutaciones en SLC40A1 con ganancia de función |
Actividad disminuida de ferroportina por mutaciones en SLC40A1 con pérdida de función |
||
|
Egreso irrestricto de hierro desde enterocitos y macrófagos hacia el plasma |
Retención de hierro en macrófagos (y enterocitos, que se descaman) |
||
|
Aumento consiguiente de la ferremia y del porcentaje de saturación de la transferrina |
Ferremia baja y porcentaje de saturación de la transferrina bajo, normal o alto según la edad del paciente |
||
|
Incremento ulterior de la ferritina sérica, reflejo de los depósitos de hierro a nivel parenquimatoso |
Hiperferritinemia marcada +++ (no tiene valor predictivo como indicador de depósitos parenquimatosos de hierro) |
||
|
Depósito de hierro a nivel de células parenquimatosas (hepatocitos, miocardiocitos, etc.) |
Depósito de hierro en macrófagos (SRE) Poco depósito de hierro (secundario) a nivel parenquimatoso |
||
|
Hepcidinemia baja (aumentada en caso de mutaciones en SLC40A1 con ganancia de función) |
Hepcidinemia elevada |
||
|
PBH |
- hepatocitos llenos de hierro - células de Kupffer vacías de hierro |
PBH |
- células de Kupffer llenas de hierro - hepatocitos con menor cantidad de hierro que las células de Kupffer |
|
RNM |
- hígado negro (por hepatocitos llenos de hierro) - bazo blanco (por macrófagos vacíos de hierro) |
RNM |
- hígado gris (por hepatocitos con depósito moderado de hierro) - bazo negro (por macrófagos muy llenos de hierro) |
|
Franca mejoría con sangrías terapéuticas perfectamente controladas |
Franca tendencia a la anemia ferropénica en caso de sangrías terapéuticas frecuentes (semejanza con IRIDA) |
||
|
Herencia autosómica: - recesiva (con baja penetrancia en caso de mutaciones HFE) - dominante en la hemocromatosis por ferroportina |
Herencia autosómica dominante |
||
3.2.1.1.1. Sobrecarga de hierro primaria hemocromatósica HFE
La hemocromatosis HFE es, seguramente, la más leve de las hemocromatosis y la única con penetrancia baja. Penetrancia baja significa que el defecto genético no es sinónimo estricto de patología clínica: muchos pacientes pueden llegar a morirse de viejos sin padecer el menor trastorno por su defecto genético... de no cruzarse en la vida con factores desencadenantes o agravantes. Ejemplos de patología con penetrancia baja son la deficiencia de G6PD clase B, la porfiria intermitente aguda, la porfiria cutánea tarda y la hemocromatosis HFE. En las dos primeras el rayo en medio de un día de sol puede ser provocado, respectivamente, por situaciones oxidantes (drogas, infecciones) -deficiencia G6PD- o activantes de la ALA sintasa-2 -PIA-. En las dos últimas, una predisposición genética suele requerir la contribución de factores adquiridos para que una enzima (UROD) o un metabolito (hierro), respectivamente, alcancen un nivel crítico (mínimo de actividad / máximo de acumulación) para deparar el cuadro clínico. La porfiria cutánea tarda es un caso extremo, ya que el defecto genético no sólo no es suficiente, sino que suele ni ser necesario para deparar cuadro clínico: de hecho, es una patología más frecuentemente adquirida que hereditaria (75 vs. 25% de los casos). En la hemocromatosis HFE el defecto genético suele no ser suficiente para generar una sobrecarga de hierro bioquímica (estadío 1) o clínica (estadío 2), pero sí es necesario para deparar el patrón de sobrecarga de hierro típico, detalle de extrema importancia diagnóstica: aumento primario del porcentaje de saturación de la transferrina vs secundario de la ferritinemia, hígado/hepatocitos llenos de hierro vs bazo/macrófagos y células de Kupffer vacíos de hierro, características típicas de una sobrecarga hemocromatósica.
Genéticamente las mutaciones en el gen HFE podemos clasificarlas en 2 categorías genotípicas: leves y severas (Tabla 5):
Tabla 5. Genotipos HFE
|
Tabla 5. Genotipos HFE |
|
|
Genotipos leves |
Genotipos severos |
|
HFE H63D |
HFE C282Y |
|
HFE S65C |
HFE con mutación que depare genotipo0 |
|
|
HFE delecionado |
La mutación HFE H63D es de alta frecuencia génica y distribución universal. La mutación HFE S65C es de baja frecuencia, descrita en el área mediterránea. Tres son los genotipos severos descritos:
- HFE C282Y, con una frecuencia génica muy alta en el noroeste europeo, pero decreciente a medida que nos acercamos a regiones mediterráneas.
- HFE con mutación que depare genotipo0 (por ejemplo: HFE R74X), muy raras,
- HFE delecionado, la causa más frecuente de hemocromatosis en la isla de Cerdeña.
Un pedido de estudio de las mutaciones del gen HFE implica investigar habitualmente la presencia o no de las mutaciones C282Y y H63D. Algunos laboratorios también incluyen la mutación S65C.
Para que un paciente tenga posibilidad de padecer un cuadro clínico hemocromatósico se requiere la presencia bialélica, en cualquier tipo de combinación, de dos defectos severos.
3.2.1.1.1.1. Sobrecarga de hierro primaria hemocromatósica HFE C282Y bialélico
O sea que, de todas las combinaciones posibles de resultados de las mutaciones estudiadas habitualmente, sólo la condición HFE C282Y/C282Y es confirmatoria de hemocromatosis. Los demás resultados posibles los voy a comentar en la sección siguiente.
Confirmado el diagnóstico de hemocromatosis, no queda más que estudiar la extensión de la sobrecarga así como sus eventuales complicaciones, e iniciar el tratamiento quelante con sangrías.
Con respecto al estudio familiar, ambos padres y todos los hijos van a ser, por lo menos, heterocigotas obligados. O sea que es muy poco probable que un estudio genético de los mismos nos depare sorpresas. Además, como ocurre en los manicomios, ni son todos los que están ni están todos los que son:
a) dada la baja penetrancia, lo más probable es que un paciente con la mutación HFE C282Y bialélica no llegue a tener problema bioquímico (y mucho menos clínico) en toda su vida, y
b) un paciente sin presencia de las mutaciones HFE C282Y ni H63D bien puede llegar a tener sobrecarga de hierro por otras causas genéticas o adquiridas.
Dada que la hemocromatosis HFE C282Y/C282Y es una hemocromatosis del adulto, de muy lenta progresión, alcanza con un control del porcentaje de saturación de la transferrina cada ¿3-5? años para detectar una sobrecarga de hierro hemocromatósica en estadío I (bioquímico), antes de que depare trastornos clínicos (estadío 2), con indicación, ahora sí, de iniciar el tratamiento que corresponda, independientemente de cuál sea el resultado del estudio genético.
Con respecto a los hermanos, según Mendel el 50% debe ser heterocigota, el 25% normal y el 25% homocigota: el propósito ya lo es, pero bien puede serlo también otro hermano.
Pero la última palabra la tiene, siempre, el porcentaje de saturación de la transferrina. El estudio genético anormal o normal en los familiares del propósito puede generar tanto ansiedad desmedida como sensación de falsa seguridad, respectivamente.
3.2.1.1.1.2 Sobrecarga de hierro primaria hemocromatósica no HFE C282 bialélico
Tengamos siempre presente que la presencia bialélica de genotipos HFE severos (C282Y/C282Y) depara una patología de baja penetrancia: sólo algunos van a alcanzar estadío bioquímico (1) y sólo unos pocos estadío clínico (2). Los porcentajes respectivos referidos en la literatura son de gran variabilidad, seguramente por diferencias en las metodologías utilizadas para calcularlos.
Cualquier otra combinación de genotipos severos o leves va a tener una penetrancia aún mucho menor, a tal punto que no va a ser capaz de generar cuadro de sobrecarga de hierro sin el "aporte" de algún otro factor, hereditario o adquirido, que contribuya a la sobrecarga de hierro.
Y lo habitual en estos casos es que el factor contributivo llegue a tener, en sí mismo, más relevancia clínica que el defecto encontrado en el gen HFE. Incluso, hoy por hoy se considera que al paciente doble heterocigota para las mutaciones HFE C282Y y H63D corresponde rotularlo como portador de una sobrecarga de hierro "de causa genéticamente no definida", antes que ponerle el rótulo de "hemocromatósico". La razón es que, seguramente, hay otras causas, además de la genética encontrada, que contribuyen a la sobrecarga de hierro y que ameritan ser investigadas y corregidas según corresponda. De no hacerlo, estamos dejando sin diagnosticar toda una parte de la patología del paciente, tal vez clínicamente más relevante que el defecto genético encontrado en sí.
Este mismo concepto se aplica, y con mayor criterio, en los pacientes con presencia sólo de las mutaciones leves H63D y/o S65C en forma mono o bialélica. Aquí el aporte genético a la sobrecarga de hierro es mínimo, y el mayor peso recae en otros factores, de evaluación imprescindible. Por este motivo algunos autores, ante la sospecha clínica de un cuadro hemocromatósico, proponen investigar sólo la presencia/ausencia de la mutación HFE C282Y: en ausencia de ésta el diagnóstico de hemocromatosis queda descartado, su presencia bialélica la confirma y su presencia monoalélica debe hacer sospechar la co-existencia de un par de factores contribuyentes a evaluar. Estos conceptos están resumidos en la tabla 6.
Tabla 6. Sobrecarga de hierro en pacientes con defectos sólo en genes hemocromatósicos
|
Tabla 6. Sobrecarga de hierro en pacientes con defectos sólo en genes hemocromatósicos |
||
|
Factores hereditarios |
+ Factores adquiridos |
|
|
defectos bialélicos en HAMP o HJV |
hemocromatosis juvenil |
apenas una o dos décadas de vida |
|
defectos bialélicos en TFR2 |
hemocromatosis del adulto |
unas 3-5 décadas de vida en el varón o 5-7 en la mujer |
|
defectos bialélicos severos en HFE |
unas 3-5 décadas de vida en el varón o 5-7 en la mujer más algún factor adquirido |
|
|
defectos bialélicos mixtos (severo y leve) en HFE (típicamente doble heterocigota C282Y y H63D) |
muchas décadas de vida y un par de factores adquiridos |
|
|
defectos bialélicos leves en HFE |
muchas décadas de vida y varios factores adquiridos |
|
|
defecto monoalélico leve en HFE |
muchas décadas de vida y muchos factores adquiridos |
|
El sombreado amarillo resalta el peso de los factores en cada platillo de la balanza.
- En las tres primeras situaciones, los factores adquiridos (más allá de la inexorable acumulación del hierro con el paso del tiempo) son nulos o poco importantes, y el patrón de depósito de hierro es típicamente hemocromatósico.
- En las dos últimas la contribución genética es escasa, el patrón de depósito de hierro NO es hemocromatósico y, lo más importante, la conducta médica hay que centrarla prioritaria, si no exclusivamente, en el diagnóstico y tratamiento de los factores adquiridos.
- En la cuarta situación ambos platillos están más o menos equilibrados, el patrón de hierro puede ser más o menos hemocromatósico, pero la contribución de los factores adquiridos es suficientemente importante como para investigarlos y no quedarse sentado echándole toda la culpa a los defectos genéticos.
Frente a un paciente con perfil de hierro sugestivo de hemocromatosis, pero con un estudio genético no HFE C282Y/C282Y, debemos plantearnos, en primer lugar y antes de seguir avanzando en el estudio de otros factores contribuyentes, qué tan "hemocromatósico" o no es realmente el perfil de nuestro paciente. Y aquí me remito a la columna de la izquierda de la tabla 4, en particular a la resonancia nuclear magnética de abdomen, con el franco contraste en el hígado (negro), desbordante de hierro, y el bazo (blanco), vacío de hierro.
Si todos los criterios convergen hacia la presencia de un componente hemocromatósico, la búsqueda debe proseguirse por el lado de los genes hemocromatósicos. Caso contrario, frente a un perfil hemocromatósico medio diluido en sus características, uno se orienta más hacia la investigación de factores adquiridos, básicamente varias de las patologías enumeradas en la tabla 3, que pueden llegar a determinar sobrecargas de hierro no hemocromatósicas.
Si todo, hasta la resonancia nuclear magnética, nos asegura que estamos realmente frente a un cuadro hemocromatósico, la investigación etiológica debemos proseguirla, como dijimos, dentro del marco de los genes hemocromatósicos, HFE o no HFE. De los no HFE voy a hablar en la sección siguiente, pero nos quedaron en el tintero otros defectos HFE severos no estudiados.
Hay un par de defectos mutacionales descritos que deparan una expresión nula del gen (genotipos 0), muy raros, que probablemente sólo vayamos a descubrir cuando tiremos la toalla y pidamos el secuenciamiento exónico de un panel de genes hemocromatósicos.
Pero hay un defecto severo, la deleción total del gen HFE (y regiones aledañas), que es la causa más frecuente de hemocromatosis en la isla de Cerdeña, con una frecuencia génica calculada de 1.01% (4 pacientes heterocigotas sobre 198 estudiados). Este defecto es traicionero, porque en una secuenciación del gen HFE la deleción heterocigota se confunde con una persona HFE homocigota normal, mientras que en condición doble heterocigota con una mutación HFE C282Y se confunde con un paciente HFE C282Y homocigota. La ausencia de polimorfismos puede hacerlo sospechar, el estudio familiar aclara el panorama y un MPLA (Multiplex ligation-dependent probe amplification) lo confirma(22).
Nada se pierde interrogando acerca de antecedentes sardos y de eventual consanguinidad, aunque sea en varias generaciones previas. Aunque complicaciones y tratamiento no difieren de otras formas de hemocromatosis, la confirmación genética de esta deleción es importante a la hora del consejo genético, por las confusiones diagnósticas que pueden surgir al momento de la secuenciación del gen HFE.
3.2.1.1.2 Sobrecarga de hierro primaria hemocromatósica no HFE
Nos quedan las mutaciones en genes hemocromatósicos no HFE para justificar el cuadro hemocromatósico franco (perfil de hierro Y resonancia nuclear magnética típicos) de nuestro paciente.
Excluido el gen HFE, cuatro genes integran este grupo. Defectos en dos de ellos cursan con cuadro clínico de hemocromatosis del adulto (TFR2 y SLC40A1), y defectos en los otros dos, con cuadro clínico de hemocromatosis juvenil (HJV y HAMP). Fisiopatológicamente la diferencia entre ambas radica sólo en la velocidad de acumulación del hierro, con requerimiento de mayor o menor tiempo para alcanzar estadíos bioquímico (1) o clínico (2).
El cuadro clínico de las hemocromatosis del adulto por TFR2 o SLC40A1 es casi idéntico al de la hemocromatosis HFE, pero con penetrancia alta, por lo que, a diferencia de la hemocromatosis HFE, no requieren de factores adquiridos para alcanzar significancia bioquímica o clínica. La hemocromatosis por ferroportina, aunque rara, tienen dos detalles particulares: es la única hemocromatosis de herencia autosómica dominante y la única que cursa con hiperhepcidinemia.
Las hemocromatosis juveniles por HJV (rara) o SLC40A1 (muy rara) son fáciles de sospechar por la edad temprana de su comienzo (segunda década de vida), y dos diferencias clínicas con la hemocromatosis del adulto:
- igual frecuencia en ambos sexos, ya que el cuadro clínico se instala antes de que la menstruación o embarazos hayan tenido tiempo de proteger a las mujeres demorando la sobrecarga, y
- mayor frecuencia de compromiso cardíaco y endócrino que en las formas del adulto, tan sólo porque estos tejidos tienen tolerancia mucho menor que el hepático a la sobrecarga excesiva de hierro y, por ende, dan manifestaciones clínicas más precozmente.
3.2.1.2 Sobrecarga de hierro primaria no hemocromatósica
Capítulo difícil, aunque de fácil sospecha si se analizan con detención los datos de laboratorio, ya que se trata de patologías de (muy) baja frecuencia y con perfiles de hierro no típicos y eventualmente variables a lo largo de la evolución del paciente, por lo que hay que ser precisos en su semiología (Tabla 7). La frecuencia de estas patologías va disminuyendo de arriba abajo: más bien raras la enfermedad por ferroportina y la aceruloplasminemia, muy raras las demás.
Tabla 7. Sobrecargas de hierro primarias no hemocromatósicas.
|
Tabla 7. Sobrecargas de hierro primarias no hemocromatósicas |
||||||||||
|
Patología |
Gen |
Herencia |
Hb |
VCM |
GR |
Reticulocitos |
Ferremia |
% Sat Tf |
Ferritina |
Hepcidina |
|
Enfermedad por ferroportina |
SLC40A1 |
AD |
= ¯ |
= ¯ |
|
¯ |
¯ |
¯ = |
|
¯ |
|
Aceruloplasminemia |
CP |
AR |
¯ |
¯ |
¯ |
¯ |
¯¯ |
¯ |
|
- |
|
Hipotransferrinemia |
TF |
AR |
¯ |
¯¯ |
¯ |
¯ |
¯¯¯ |
100% |
= |
¯ |
|
Deficiencia Steap3 |
STEAP3 |
AR |
¯¯ |
¯ |
¯ |
¯ |
|
|
|
|
|
Deficiencia DMT1 |
SLC11A2 |
AR |
¯¯ |
¯¯¯ |
¯ |
¯ |
|
|
|
|
Todas estas patologías cursan con anemia microcítica hiporregenerativa hierro restricta (sombreado en gris), la enfermedad con ferroportina generalmente sólo después del inicio de un tratamiento quelante con sangrías a dosis relativamente altas. La combinación de anemia microcítica hierro-restricta con una sobrecarga sistémica de hierro, sugiere claramente algún defecto en la utilización del hierro. Justamente lo más importante a destacar en esta tabla (sombreado en rosa y verde) es la heterogeneidad del perfil de hierro.
Las proteínas involucradas integran el elenco responsable del tránsito del hierro desde su absorción intestinal hasta su destino citoplasmático final, y en este orden están mostradas en la tabla. En las dos primeras patologías el problema radica en el ingreso del hierro al plasma, en la tercera en su transporte y en las dos últimas en su egreso del plasma. El gran ausente en esta tabla es el receptor de transferrina-1, cuya deficiencia, muy poco frecuente, depara cuadro clínico de inmunodeficiencia combinada relacionada a TFRC (gen que codifica para el receptor de transferrina-1)(23).
3.2.1.2.1 Sobrecarga de hierro primaria no hemocromatósica. Enfermedad por ferroportina
Proteína transmembrana (con 12 hélices transmembrana, 6 segmentos extracelulares y 5 segmentos intracelulares) ubicada en la cara plasmática de varias células (enterocitos, macrófagos, hepatocitos, eritroblastos -con la función de reciclar el hierro excedente de la síntesis del hemo-, trofoblastos placentarios, y, en grado mínimo, algunas otras), la ferroportina recibe órdenes divergentes: aperturistas por parte del hierro ferroso intracelular, obstruccionistas y degradantes por parte de la hepcidina plasmática. Así es como, groseramente, mutaciones en residuos que miran hacia el interior de la célula suelen afectar su relación con el hierro, mientras que mutaciones en los que miran hacia el exterior tienden a afectar su relación con la hepcidina. Y funcionalmente pueden deparar una ganancia o una pérdida de su función exportadora de hierro, y/o una conservación o pérdida de su función como receptora de la hepcidina. La relación genotipo-fenotipo es bastante compleja, y así lo demuestra una revisión(24) de 359 pacientes con 60 variantes distintas de ferroportina y cuadro clínicos de hemocromatosis por ferroportina o de enfermedad por ferroportina, en general, pero no siempre, relacionados con mutaciones con ganancia o con pérdida de función respectivamente (Tabla 8).
Tabla 8. Ferroportina. Mutaciones con funcionalidad y respuesta a la hepcidina diferentes(24)
|
Tabla 8. Ferroportina. Mutaciones con funcionalidad y respuesta a la hepcidina diferentes(24) |
|||
|
Funcionalidad |
Respuesta a hepcidina |
Nº variantes |
Nº pacientes |
|
Variantes con ganancia de función |
resistente |
7 |
37 |
|
sensible |
7 |
12 |
|
|
conflictiva, incierta o desconocida |
13 |
61 |
|
|
Variantes con pérdida de función |
resistente |
5 |
79 |
|
sensible |
4 |
57 |
|
|
conflictiva, incierta o desconocida |
12 |
80 |
|
|
Variantes no clasificadas |
12 |
33 |
|
Mientras la hemocromatosis por ferroportina es clínicamente casi idéntica a la hemocromatosis por HFE -por eso figura dentro del capítulo de las hemocromatosis no HFE-, la enfermedad por ferroportina, su antítesis, es la variante más común de las sobrecargas de hierro primarias no hemocromatósicas.
En la hemocromatosis por ferroportina (como en cualquier hemocromatosis) la acumulación de hierro es primariamente en el parénquima (hepático, pancreático, cardíaco, en este orden cronológico), con elevación secundaria de la ferritinemia. En la enfermedad por ferroportina, a la inversa, la acumulación es primariamente en los macrófagos y, secundariamente en los tejidos parenquimatosos.
Ambos efectos secundarios son difíciles de explicar:
- la hiperferritinemia de las hemocromatosis debe significar que la ferritinemia refleja (también) los depósitos de hierro mas allá de los macrofágicos, que están típicamente vacíos.
- la sobrecarga parenquimatosa tardía en la enfermedad por ferroportina (hígado y bazo oscuros) es más difícil de explicar con nuestros conocimientos fisiopatológicos actuales.
La enfermedad por ferroportina también es fisiopatológicamente parecida, pero clínicamente antagónica, de la anemia ferropénica refractaria al hierro (IRIDA, por sus siglas en inglés). En ambas hay una deficiencia en la función exportadora del hierro, primaria en la enfermedad por ferroportina, secundaria a un exceso de hepcidina por mutaciones en el gen de la matriptasa (TMPRSS6) en IRIDA. Pero las consecuencias no pueden ser más opuestas: sobrecarga de hierro en una, anemia ferropénica en la otra.
El patrón de herencia de las ferroportinopatías (hemocromatosis y enfermedad) es dominante, muy posiblemente por efecto dominante negativo y no por haploinsuficiencia: se postula que, al menos en algunos tejidos, la ferroportina funciona en forma tetramérica, requiriendo la normalidad estructural de todas sus unidades para ser funcional. Esto coincide con que no hay descritas ferroportinopatías por defectos que determinen genotipos 0 (parecería que son más "nocivos" los genotipos X que los 0, con un remanente ferroportínico normal del 20 vs. 50% respectivamente), e implica que siempre hay un resto de ferroportina funcionante en la enfermedad por ferroportina. Por el contrario, en la IRIDA la ferroportina está totalmente inhibida por la hiperhepcidinemia. Así, la "ausencia" de ferroportina en la IRIDA afecta tanto la salida del hierro desde los macrófagos como desde los enterocitos (con ferropenia consecuente y resistencia al tratamiento con hierro), mientras que en la enfermedad por ferroportina la deficiencia afecta fundamentalmente a los macrófagos (por los que pasan unos 20 mg diarios de hierro proveniente de la fagocitosis y degradación de los hematíes senescentes) y no tanto a los enterocitos (que sólo exportan hacia el plasma 1 mg diario de hierro en el varón adulto y 2 mg en la mujer en edad fértil). O sea que en la enfermedad por ferroportina el hierro queda retenido en macrófagos (la poca ferroportina no da abasto a cumplir su función), con la consecuente eritropoyesis hierro restricta, aumento de la liberación de eritroferrona, menor síntesis de hepcidina, mayor absorción del hierro en enterocitos (la poca ferroportina alcanza para cumplir su función) y sobrecarga orgánica de hierro.
Esta eritropoyesis hierro restricta por secuestro del hierro en los macrófagos (paralelismo con la anemia de los procesos inflamatorios) explica la intolerancia y rápida anemización si las dosis de sangrías indicadas para paliar la sobrecarga de hierro son semejantes a las que uno usaría en pacientes con hemocromatosis. La sospecha de la enfermedad por ferroportina surge a partir de una hiperferritinemia franca con una ferremia baja y un porcentaje de saturación de la transferrina variable según la edad del paciente, y con patrón de herencia autosómica dominante. La RNM de abdomen es la clave orientadora, con un bazo negro por el hierro que no puede salir de los macrófagos por el defecto de la ferroportina, y un hígado, si no blanco, al menos más claro que el bazo, y que se blanquea rápidamente (y antes que el bazo) a partir del inicio del tratamiento quelante con sangrías. Si, por algún otro motivo, se tiene acceso a una punción biopsia hepática, la detección de los depósitos de hierro a nivel de las células de Kupffer y no de los hepatocitos no hace más que confirmar lo ya sabido. Con o sin confirmación genética, el cuadro típico es suficiente para iniciar sangrías a dosis bajas y controlando muy de cerca los valores e índices eritrocíticos, dada la fácil anemización por la incapacidad del hierro de salir de sus depósitos para acceder a los eritroblastos.
3.2.1.2.2 Sobrecarga de hierro primaria no hemocromatósica. Aceruloplasminemia
El problema aquí radica en la imposibilidad del hierro ferroso, tal como es exportado desde el citoplasma por la ferroportina, de ser oxidado a férrico para poder ser captado por la transferrina. En oposición a la hepcidina (desestabilizadora de la ferroportina), la ceruloplasmina actúa como su estabilizadora, por lo que en la aceruloplasminemia disminuye la expresión en membrana de la ferroportina. Por eso el perfil de hierro es discordante, con ferremia y porcentaje de saturación de la transferrina sugerentes de componente ferropénico, pero con ferritinemia elevada, no como reactante de fase aguda como podría serlo en una anemia de los procesos inflamatorios, sino reflejando un secuestro del hierro, dificultado para salir de los macrófagos, pero no de los enterocitos, donde la oxidación del hierro la cumple la hefestina. De la hefestina no se conocen disfunciones, seguramente porque la ceruloplasmina la cubre en caso de deficiencia. El perfil de hierro en la aceruloplasminemia es más o menos parecido al de la enfermedad por ferroportina, sólo que el porcentaje de saturación de transferrina está siempre bajo en aquélla, mientras que en ésta suele estar bajo en etapas iniciales y aumentado en etapas avanzadas, en paralelo con la ferritinemia.
Pero en la aceruloplasminemia el cuadro clínico con el paso del tiempo excede lo hematológico:
- hacia los 20-30 años suele hacerse evidente el depósito de hierro a nivel esplácnico, con un patrón imagenológico de hierro semejante al hemocromatósico: más en hígado que en bazo, aunque en un grado más leve, por lo que la cirrosis es rara.
- hacia los 30-40 años se instala una diabetes insulino-dependiente, más por estrés oxidante de las células b, con apoptosis consecuente, que por depósito de hierro, que es mayor en el páncreas exócrino que en el endócrino.
- y hacia los 40-50 años comienzan los trastornos neurológicos (ataxia cerebelosa, movimientos involuntarios, parkinsonismo, trastornos de conducta). En Japón, donde la aceruloplasminemia es más frecuente que la hemocromatosis, una complicación común es la degeneración retiniana. La ceruloplasmina, en particular como proteína anclada a través del puente glicolipídico, juega un rol especial en el egreso del hierro desde las células epiteliales del plexo coroideo, glía y astrocitos hacia el líquido cefalorraquídeo. A nivel histológico la acumulación de hierro es mayor en estas células que en las neuronas, y a nivel anatómico el depósito predomina a nivel de ganglios basales y cerebelo(25).
La sospecha de aceruloplasminemia puede surgir a partir de la triada anemia microcítica + niveles bajos de ferremia y porcentaje de saturación de la transferrina + ferritinemia elevada. Si la elevación de la ferritinemia no es significativa, la medición de la concentración de hierro hepático va a confirmar la sobrecarga de hierro. El perfil de hierro es sugestivo de patología hierro restricta, pero con mayor severidad y cronicidad que una anemia de los procesos inflamatorios. Si el paciente es adulto, la presencia de diabetes o signos de neuropatía extrapiramidal incrementan la sospecha. El cuadro neurológico puede hacer confundir con la enfermedad de Wilson, pero aquí la sobrecarga es de cobre (no de hierro).
Una ceruloplasminemia baja orienta francamente el diagnóstico (más baja que en la enfermedad de Wilson), acompañada de una cupremia baja, ya que la ceruloplasmina es la transportadora del 95% del cobre plasmático.
La detección de un defecto patogénico en el gen CP confirma el diagnóstico, aunque no es imprescindible para ponerle el rótulo al paciente si el cuadro clínico es típico. Sólo queda iniciar tratamiento con agentes antioxidantes y terapia de remplazo enzimático (plasma fresco congelado por vía endovenosa o peritoneal, con cruce de la barrera hematoencefálica por la ceruloplasmina aportada). El tratamiento quelante de hierro es de efectividad muy dudosa.
3.2.1.2.3 Sobrecarga de hierro primaria no hemocromatósica. Hipotransferrinemia
En la hipotransferrinemia (la atransferrinemia sería prácticamente incompatible con la vida) la poca transferrina circulante transporta una cantidad muy escasa de hierro (ferremia muy baja, en contraste con las deficiencias de Steap3 y DMT1) pese a estar saturada al 100% (por descenso franco del denominador). La anemia es también típicamente hierro-restricta, pero la ferritinemia no suele estar muy aumentada.
3.2.1.2.4 Sobrecarga de hierro primaria no hemocromatósica. Deficiencias de Steap3 o DMT1
El transportador de metales divalentes, responsable del ingreso del hierro a las células, cumple dos papeles en este drama, en ambos casos determinando que el hierro divalente (ferroso) cruce la membrana hacia al citoplasma desde: a) la luz intestinal, en el enterocito, o b) el endosoma que invagina a la transferrina diférrica unida al receptor de transferrina-1, en cualquier célula del organismo. ¿Cuál de esos dos roles es el más afectado en caso de mutaciones con pérdida de función? Aparentemente sólo el segundo, sugiriendo que puede haber otros mecanismos no conocidos que permiten la captación duodenal del hierro desde la luz del tubo digestivo. Por eso en la tabla 7 lo he ubicado sólo al final de la lista, ya llegando el hierro a su destino metabólico final.
El perfil de hierro es idéntico en las deficiencias de DMT1 y de Steap3 (la proteína encargada en el endosoma de reducir a ferroso el hierro férrico liberado, acidificación ATP dependiente mediante, desde la transferrina diférrica unida a su receptor -TfR1-): aumento de la ferremia y, sobre todo, del porcentaje de saturación de la transferrina, fiel reflejo de la imposibilidad del hierro circulante de cruzar la membrana plasmática e ingresar a las células. En los 3 miembros de la única familia descrita con deficiencia de Steap3 se han detectado sideroblastos en anillo. Mientras que sistémicamente hay una sobrecarga de hierro, la anemia en estos pacientes es hierro-restricta, por lo que se agrava con tratamiento quelante, mientras que mejora parcialmente con eritropoyetina.
3.2.2 Sobrecargas de hierro secundarias
El subcapítulo de las sobrecargas de hierro secundarias focalizadas excede en varios casos el feudo hematológico, pero al que tal vez debamos asomarnos en el marco de los diagnósticos diferenciales. Las anemias sideroblásticas hereditarias figuran tanto dentro de las sistémicas (donde realmente pertenecen) y de las focalizadas (sólo por la localización patognomónica y diagnóstica del depósito férrico a nivel intramitocondrial). La anemia de los procesos inflamatorios (con o sin descenso de los valores eritrocíticos, pero con perfil de hierro típico) no es más que un secuestro retículo endotelial preventivo del hierro, con anemia "hierro restricta" consecuente (aunque la causa real del descenso de los valores eritrocíticos en esta patología es una deficiencia relativa de eritropoyetina). En la neuroferritinopatía se combina una hipoferritinemia moderada con un cuadro neurológico, que contrasta con la combinación trastorno neurológico + hiperferritinemia, típica de la aceruloplasminemia hereditaria o con el cuadro de hemocromatosis juvenil y alteraciones neurológicas en tres pacientes con mutaciones constitucionales en el gen PIGA que impiden el anclaje en membrana de proteínas tales como la hemojuvelina y la ceruloplasmina(19).
Con respecto a las sobrecargas de hierro secundarias sistémicas, su común denominador es el ingreso excesivo de hierro al organismo, ya sea por alteración secundaria del mecanismo regulador de la absorción enteral y/o por su puenteo parenteral (medicamentoso o transfusional). En ambos casos el depósito de hierro es típicamente no hemocromatósico, con a) acumulación semejante a nivel parenquimatoso-hepático que retículo endotelial-esplénico (en caso de ingreso enteral del hierro), o b) predominantemente retículo endotelial-esplénico -con cierta semejanza al patrón ferroportínico y opuesto al hemocromatósico- (en caso de ingreso parenteral del hierro -transfusiones-).
El ejemplo típico de esta "discordancia" son las talasemias transfusión vs. no transfusión dependientes. En las primeras el destino final de los hematíes transfundidos es la fagocitosis macrofágica a nivel esplénico, reflejado nítidamente en los niveles de ferritinemia. Aquí los depósitos hepáticos de hierro son sólo un reflejo secundario de la sobrecarga sistémica.
Pero en las talasemias no transfusión dependientes (beta-talasemia intermedia, enfermedad con hemoglobina H, incluso beta-talasemia mayor antes del inicio del régimen transfusional regular, deficiencia de piruvato quinasa, etc.) la absorción incrementada del hierro a nivel enteral está activada desde la eritropoyesis inefectiva a través de la eritroferrona que, capturando las proteínas morfogénicas óseas-6 y -2 en el espacio de Disse hepático, evita que éstas induzcan la transcripción del gen de la hepcidina. Así la ferroportina queda en libertad de acción para volcar hacia el plasma el hierro contenido en las células que la expresan: enterocitos y macrófagos. En las talasemias no transfusión dependientes, entonces, el hepatocito es la primera estación del hierro absorbido a nivel intestinal, que sólo secundariamente va a ir depositándose en los macrófagos. Por ende, la ferritinemia, que podrá reflejar bien el hierro depositado a nivel macrofágico, pero no tanto a nivel parenquimatoso, en este caso subvalora la cantidad de hierro depositada en hígado. De aquí surge la recomendación de iniciar tratamiento quelante con ferritinemias mayores a 1.000 ug/L en los pacientes con talasemias transfusión dependientes, pero mayores a 800 ug/L en los no transfusión dependientes.
Este concepto también es válido para las demás patologías con eritropoyesis inefectiva (hereditaria o adquirida -mielodisplasias-), pero en ninguna de ellas está indicado un régimen transfusional regular con el objetivo de frenar la hematopoyesis patológica del paciente. Al ser la sobrecarga de hierro más absortiva que transfusional, en estas patologías es preferible apoyarse más en la concentración hepática de hierro que en la ferritinemia para la evaluación de la sobrecarga de hierro y la decisión terapéutica.
En la hepatopatía gestacional autoinmune, patología familiar pero no heredable (comparémosla con la anemia hemolítica del recién nacido), el problema es la agresión desvastadora de anticuerpos maternos (IgG activantes de complemento) contra antígenos hepáticos fetales que la madre no reconoce como propios, con la incapacidad consecuente del hígado fetal de sintetizar hepcidina, transferrina, angiotensina, etc., etc., generando una sobrecarga de hierro "hemocromatósica" severa (aunque no típica, porque el defecto proteico es múltiple, no sólo hepcidínico), antes denominada "hemocromatosis neonatal".
4. Conclusión
Muy diferentes situaciones clínicas pueden deparar distintos perfiles sugestivos de sobrecarga de hierro. El conocimiento lo más preciso posible del metabolismo del hierro nos es de gran ayuda para correlacionar fisiopatológicamente la(s) etiología(s) responsable(s) con los datos semiológicos encontrados. Por ejemplo, no corresponde rotular como "sobrecarga de hierro" una hiperferritinemia no acompañada de un aumento del porcentaje de saturación de la transferrina, ni justificar una sobrecarga de hierro por la sola presencia de una mutación HFE leve en condición mono o bialélica. Cualquiera que sea el diagnóstico final al que arribemos, indicaremos, siempre que sea posible, el tratamiento etiológico que corresponda, pero la duda suele surgir, particularmente en caso de hiperferritinemias reactivas, con respecto a la conveniencia o no de indicar tratamiento sintomático (quelante). En este sentido cada situación clínica debe ser evaluada particularmente, sin perder de vista si la sobrecarga de hierro es de causa primaria o consecuencia secundaria de otra patología de base.
Bibliografía
1. Fonseca Ó, Ramos AS, Gomes LTS y col. New Perspectives on Circulating Ferritin: Its Role in Health and Disease. Molecules. 2023 Nov 22;28(23):7707. doi: 10.3390/molecules28237707.
2. Cohen LA, Gutierrez L, Weiss A y col. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood. 2010 Sep 2;116(9):1574-84. doi: 10.1182/blood-2009-11-253815.
3. Wang W, Knovich MA, Coffman LG y col. Serum ferritin: Past, present and future. Biochim Biophys Acta. 2010 Aug;1800(8):760-9. doi: 10.1016/j.bbagen.2010.03.011.
4. Curtis AR, Fey C, Morris CM y col. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001 Aug;28(4):350-4. doi: 10.1038/ng571.
5. Vidal R, Ghetti B, Takao M y col. Intracellular ferritin accumulation in neural and extraneural tissue characterizes a neurodegenerative disease associated with a mutation in the ferritin light polypeptide gene. J Neuropathol Exp Neurol. 2004 Apr;63(4):363-80. doi: 10.1093/jnen/63.4.363.
6. Levi S, Cozzi A, Arosio P. Neuroferritinopathy: a neurodegenerative disorder associated with L-ferritin mutation. Best Pract Res Clin Haematol. 2005 Jun;18(2):265-76. doi: 10.1016/j.beha.2004.08.021.
7. Lorcerie B, Audia S, Samson M y col. Diagnosis of hyperferritinemia in routine clinical practice. Presse Med. 2017 Dec;46(12 Pt 2):e329-e338. doi: 10.1016/j.lpm.2017.09.028.
8. Hearnshaw S, Thompson NP, McGill A. The epidemiology of hyperferritinaemia. World J Gastroenterol. 2006 Sep 28;12(36):5866-9. doi: 10.3748/wjg.v12.i36.5866.
9. Sandnes M, Ulvik RJ, Vorland M y col. Hyperferritinemia-A Clinical Overview. J Clin Med. 2021 May 7;10(9):2008. doi: 10.3390/jcm10092008.
10. Camaschella C, Poggiali E. Towards explaining "unexplained hyperferritinemia". Haematologica. 2009 Mar;94(3):307-9. doi: 10.3324/haematol.2008.005405.
11. Adams PC, Barton JC. A diagnostic approach to hyperferritinemia with a non-elevated transferrin saturation. J Hepatol. 2011 Aug;55(2):453-8. doi: 10.1016/j.jhep.2011.02.010.
12. Sogni P, Buffet C. Démarche clinique devant une hyperferritinémie [Clinical evaluation of a hyperferritinemia]. Presse Med. 2013 Apr;42(4 Pt 1):405-10.
13. VanWagner LB, Green RM. Elevated serum ferritin. JAMA. 2014 Aug 20;312(7):743-4. doi: 10.1001/jama.2014.302.
14. Ong SY, Nicoll AJ, Delatycki MB. How should hyperferritinaemia be investigated and managed? Eur J Intern Med. 2016 Sep;33:21-7. doi: 10.1016/j.ejim.2016.05.014.
15. Ravasi G, Pelucchi S, Mariani R y col. Unexplained isolated hyperferritinemia without iron overload. Am J Hematol. 2017 Apr;92(4):338-343. doi: 10.1002/ajh.24641.
16. Cullis JO, Fitzsimons EJ, Griffiths WJ y col; British Society for Haematology. Investigation and management of a raised serum ferritin. Br J Haematol. 2018 May;181(3):331-340. doi: 10.1111/bjh.15166.
17. Cadenas B, Fita-Torró J, Bermúdez-Cortés M y col. L-Ferritin: One Gene, Five Diseases; from Hereditary Hyperferritinemia to Hypoferritinemia-Report of New Cases. Pharmaceuticals (Basel). 2019 Jan 23;12(1):17. doi: 10.3390/ph12010017.
18. Kannengiesser C, Jouanolle AM, Hetet G y col. A new missense mutation in the L ferritin coding sequence associated with elevated levels of glycosylated ferritin in serum and absence of iron overload. Haematologica. 2009 Mar;94(3):335-9. doi: 10.3324/haematol.2008.000125.
19. Muckenthaler L, Marques O, Colucci S y col. Constitutional PIGA mutations cause a novel subtype of hemochromatosis in patients with neurologic dysfunction. Blood. 2022 Mar 3;139(9):1418-1422. doi: 10.1182/blood.2021013519.
20. Kato J, Fujikawa K, Kanda M y col. A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am J Hum Genet. 2001 Jul;69(1):191-7. doi: 10.1086/321261.
21. Girelli D, Busti F, Brissot P y col. Hemochromatosis classification: update and recommendations by the BIOIRON Society. Blood. 2022 May 19;139(20):3018-3029. doi: 10.1182/blood.2021011338.
22. https://sahcampus.org.ar/ - Cursos disponibles - Eritropatías. Casos Clínicos Comentados - Febrero 2024 - 2402-B - Sobresarda de hierro.
23. Aljohani AH, Al-Mousa H, Arnaout R y col. Clinical and Immunological Characterization of Combined Immunodeficiency Due to TFRC Mutation in Eight Patients. J Clin Immunol. 2020 Nov;40(8):1103-1110. doi: 10.1007/s10875-020-00851-1.
24. Vlasveld LT, Janssen R, Bardou-Jacquet E y col. Twenty Years of Ferroportin Disease: A Review or An Update of Published Clinical, Biochemical, Molecular, and Functional Features. Pharmaceuticals (Basel). 2019 Sep 9;12(3):132. doi: 10.3390/ph12030132.
25. Pietrangelo A. Iron and the liver. Liver Int. 2016 Jan;36 Suppl 1:116-23. doi: 10.1111/liv.13020.