Congenital hypofibrinogenemia with bone cyst: a case report with review of literature
Abstract
Congenital hypofibrinogenemia is a rare autosomal recessive condition leading to low plasma fibrinogen levels, affecting approximately one in a million. The clinical phenotype is diverse and may vary from bleeding and thrombosis to the absence of symptoms. Bone cysts are an infrequent complication of hypofibrinogenemia. This case report describes the clinical history, diagnosis, and treatment of a 13-year-old patient with congenital hypofibrinogenemia with a bone cyst.
Keywords: hypofibrinogenemia,
bone cysts,
fibrinogen deficiency.
Abbreviations
APTT: activated partial thromboplastin time
CT: computed tomography
INR: international normalized ratio
PT: prothrombin time
RBS: random blood sugar
SGOT: serum glutamic oxaloacetic transaminase
SGPT: serum glutamate pyruvate transaminase
TT: thrombin time
Case report
A 13-year-old male child presented with pain in his left arm. He had difficulty in carrying his bag to school. The patient had no history of any trauma. On examination, the child was conscious, oriented, and afebrile, and on local examination, the left arm was swollen and tender, and the range of motion was painful. No abnormalities were found in the systemic examination.
The patient's medical records were analyzed. The patient had a history of umbilical cord bleeding at birth. He was admitted for occipital hemorrhage at six months of age and diagnosed with congenital hypofibrinogenemia. He had normal development milestones. The patient was hospitalized many times for bleeding manifestations and was treated with FFP and cryoprecipitate. There was no family history of similar complaints. The need of exome sequencing was advised, however the parents had financial issues.
His laboratory parameters are summarized in table 1.
Table 1.
|
Test |
Value |
Range |
|
RBS (mg/dl) |
86 |
< 140 |
|
Urea (mg/dl) |
12 |
5-20 |
|
Creatinine (mg/dl) |
0.5 |
0-8-1.2 |
|
Total bilirubin (mg/dl) |
1.0 |
0.1-1.2 |
|
Direct bilirubin (mg/dl) |
0.4 |
0.1-0.3 |
|
SGOT (U/L) |
31 |
7-56 |
|
SGPT (U/L) |
27 |
8-45 |
|
ALP (IU/L) |
146 |
44 to 147 |
|
Total leucocyte count (cells/mm3) |
9900 |
4000-11000 |
|
Hemoglobin (g/dl) |
10.7 |
11.2 to 14.5 |
|
Platelets (103/microL) |
468 |
150-450 |
|
PT (secs) |
>180 |
11- 13.5 |
|
APTT (secs) |
>180 |
30-40 |
|
Serum fibrinogen (mg/dl) |
40 |
200 and 400 |
|
Clot retraction |
No clot formed |
0-2 hours |
|
TT (sec) |
No clot formed |
19 |
|
Factor XIII screening test |
No clot formed |
|
Viral markers were negative for HIV and hepatitis-B.
These reports suggested that the patient had a normal liver and renal function and a prolonged coagulation profile with low serum fibrinogen. Initially, he was treated with cryoprecipitate transfusion, analgesics, anti-fibrinolytics, and supplements. Rheumatology opinion was obtained. X-ray and CT screening of the left humerus were performed.
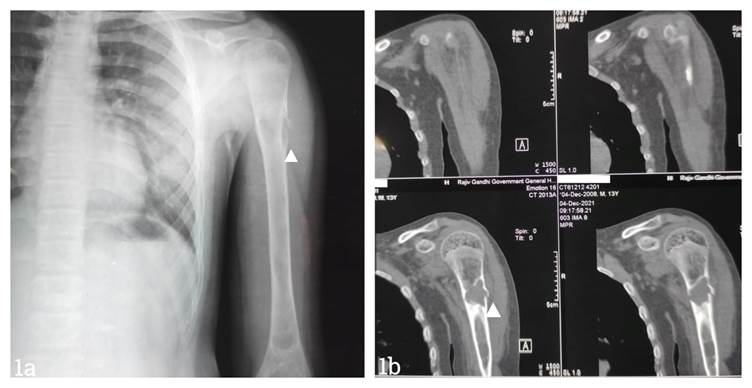
Fig 1a. X-ray showing a proximal fracture in the left humerus. Fig 1b. CT scan of the left shoulder showing a solitary bone cyst in the left humerus.
X-ray revealed a simple bone cyst in the left proximal humerus with a pathological fracture and a united right proximal humerus fracture. USG screening of the left shoulder revealed a solitary bone cyst.
CT scan of the left shoulder had a unilocular loculated central lytic expansile lesion with endosteal scalloping involving the diaphysis of the proximal humerus and a transverse fracture at the site of the lesion, with a thin, detached fracture fragment within the lesion and minimal solid periosteal reaction, suggestive of a solitary bone cyst complicated by fracture. The fracture was managed conservatively with a U slab. No other procedure was performed due to the risk of bleeding manifestations. His pain reduced, and there was no bleeding. Normal callus formation was observed in the X-ray after four weeks of management.
Discussion
Disorders of fibrinogen can be inherited or acquired. Congenital fibrinogen abnormalities, which include quantitative (afibrinogenemia, hypofibrinogenemia) and qualitative (dysfibrinogenemia, hypo dysfibrinogenemia) disorders are infrequent conditions impacting the blood coagulation process(1). The liver produces the plasma glycoprotein fibrinogen, which is involved in the last stage of the coagulation cascade. In addition, it is an acute-phase reactant, that promotes angiogenesis, fibrinolysis, and platelet aggregation, and engages in other physiologic processes(1).
Congenital forms are typically autosomal recessive. Mutations affecting any one of the three fibrinogen genes -FGA, FGB and FGG- localized on chromosome 4 have been linked. Qualitative defects are commonly the result of mutations in the gene encoding fibrinogen. A proportionate decrease in both functional and antigen fibrinogen levels is known as hypofibrinogenemia. While hypofibrinogenemia is typically asymptomatic, afibrinogenemia is frequently linked to mild-to-severe hemorrhage. The symptoms vary based on the plasma fibrinogen level. Thromboembolism may occur either spontaneously or in association with fibrinogen substitution therapy. These patients usually present in the neonatal period with a history of trauma during delivery(2). Unfortunately the genetic sequencing was not done in our case.
Table 2 . Classification of congenital quantitative fibrinogen disorders
|
Types |
Description |
|
1A. Afibrinogenemia
1B. Afibrinogenemia with thrombotic phenotype |
Afibrinogenemic patients, either with bleeding or asymptomatic individuals Afibrinogenemic with a thrombus |
|
2A. Severe hypofibrinogenemia 2B.Moderate hypofibrinogenemia 2C. Mild hypofibrinogenemia 2D. Hypofibrinogenemia with fibrinogen storage disease |
Functional fibrinogen level less than 0.5 g L-1 between 0.5 to 0.9 g L-1 between 1 g L-1 to lower limit of normal level Familial hypofibrinogenemia with the accumulation of fibrin in hepatocytes |
No clotting factors cross the placental barrier, and the fetus starts hepatic production of anticoagulants and procoagulants at the fifth week of gestation. Consequently, all clotting factors are present from birth. It is crucial to rule out secondary causes such as liver disease, hypoalbuminemia and consumptive coagulopathy before confirming the diagnosis of congenital hypo- and afibrinogenemia. In our patient, liver and renal function tests were normal, ruling out the secondary causes. We did not perform the antigenic fibrinogen study or PT or APTT mixing studies or coagulation factor dosages for the differential diagnosis with other coagulation disorders.
Affected individuals may complain of ecchymoses, subcutaneous hemorrhages or hemarthroses or exhibit no apparent clinical abnormalities(3). Previously, bone cysts have been described in a few cases as a rare complication of afibrinogenemia. Many articles have reported that juxta trabecular hemorrhages in the metaphysis lead to the formation of intraosseous cysts and cause the remodeling of bone trabeculae. Most of these individuals experience bone cysts in their long bones during childhood, which can be related to growth(4,5).
Cysts regress in certain situations, most likely due to bone remodeling. It has been proposed that trabeculae are partially absorbed during haemorrhage, and new bone develops concurrently(6). More clinical research is required to fully comprehend the pathophysiology of bone cysts in hypo- and afibrinogenemia.
The initial step in the laboratory diagnosis of fibrinogen disorders is APTT and PT, followed by a functional assessment of fibrinogen levels using the Clauss method and an antigenic measurement. If antigenic measurement is not possible, a prothrombin time-derived fibrinogen is an indirect measurement obtained from a prothrombin time curve's change in light transmission or scatter(7). Apart from the conventional coagulation tests that rely on fibrin formation, global coagulation assays help monitor the administration of replacement treatment. A crucial step in validating the clinical diagnosis is genetic testing(8).
Substitution therapy is most effective for treating bleeding episodes in patients with congenital fibrinogen disease. Fibrinogen must be replaced using cryoprecipitate, fresh frozen plasma or fibrinogen concentrate to treat bleeding episodes(9). FFP has several drawbacks, including transfusion-related hazards and a lower amount of infused fibrinogen that requires repeated administration to reach the desired level(10). The main reasons for the critical application of fibrinogen concentrates are safety, precision, ease of dosage in tiny doses, and speed of administration(11).
summarize the reported cases of afibrinogenemia with bone cysts. Eight patients had bone cysts as a complication of afibrinogenemia, and six of them had pain in their extremities. Bone cysts were primarily located in the vicinity of the cortex or trabeculae in the diaphysis of long bones, especially in the femur, tibia, and humerus(5).
Another case was reported in the Netherlands. A 16-year-old boy was diagnosed with afibrinogenemia shortly after birth. He had a history of pain in both legs since 4 and a half years of age. From the age of 9, the child experienced increasing pain in both legs. Moreover, a total body MRI also revealed cysts in the diaphysis of the humeri and femora. The child was treated prophylactically with fibrinogen concentrate, and he experienced immediate pain relief after the infusion of fibrinogen(5).
Another patient was a 10-year-old girl whose parents were Moroccan consanguineous; at the age of two, she was diagnosed with afibrinogenemia. She started experiencing leg and knee pain at the age of 4, but X-rays revealed no abnormalities. Intramural cystic lesions were observed in the diaphysis of the right humerus, femur, and left tibia during a whole-body MRI. Her bone discomfort subsided once fibrinogen concentrate was started as part of a prophylactic treatment plan(5,8).
Our patient also had a similar history and experienced reduction in pain after the initiation of fibrinogen concentrate.
Not every instance reported present with limb pain. When there is hypo- or afibrinogenemia and symptoms of "rheumatic" pain in the extremities, it could be prudent to look for bone cysts. The best diagnostic imaging modality is a whole-body MRI since it allows for the simultaneous evaluation of bone marrow and soft tissue. There are no clear clinical guidelines on the use of primary prophylaxis in patients with hypo- or afibrinogenemia. Nonetheless, it is advised that secondary prophylaxis be taken into consideration to maintain fibrinogen trough levels above 0.5 g L-1 if the patient has previously experienced potentially fatal bleeding, such as an intracranial hemorrhage with a risk of recurrence(9).
Conclusion
Hypofibrinogenemia is rare in clinical practice. Bone cysts are an infrequent complication of fibrinogen defects, especially in patients with hypofibrinogenemia. There are no guidelines for this unusual illness, and the few cases documented so far also involve individuals with afibrinogenemia. Most of the cysts are found in the diaphysis of long bones, and some of them are regressive, most likely because of bone remodeling. Patients with hypo- or afibrinogenemia who report "rheumatic" pain in their extremities may benefit from a whole-body MRI to identify bone cysts. Treatment with fibrinogen concentrate can reduce pain but has little effect on radiological degradation.
Author contributions:
VY and MM drafted the article, and both were involved in the concept and design of the article.
All the authors are solely accountable for the accuracy and integrity of every part of this work.
Declaration of patient consent.
Written informed consent was obtained from the parents for publication.
Ethical approval
The study was approved by the Madras Medical College, Ethical Committee.
financial support and sponsorship
Nil.
Conflicts of interest
There is no conflict of interest.
References
1. de Moerloose P, Casini A, Neerman-Arbez M. Congenital fibrinogen disorders: an update. Semin Thromb Hemost. 2013 Sep;39(6):585–95.
2. Casini A, Undas A, Palla R, Thachil J, de Moerloose P, Subcommittee on Factor XIII and Fibrinogen. Diagnosis and classification of congenital fibrinogen disorders: communication from the SSC of the ISTH. J Thromb Haemost. 2018 Sep;16(9):1887–90.
3. Ménaché D. Congenital abnormal fibrinogens. Prog Clin Biol Res. 1981;72:205–20.
4. Neerman-Arbez M, Casini A. Clinical Consequences and Molecular Bases of Low Fibrinogen Levels. Int J Mol Sci. 2018 Jan 8;19(1).
5. van Meegeren MER, de Rooy JWJ, Schreuder HWB, Brons PPT. Bone cysts in patients with afibrinogenaemia: a literature review and two new cases. Haemophilia. 2014 Mar;20(2):244–8.
6. Lagier R, Bouvier CA, Van Strijthem N. Skeletal changes in congenital fibrinogen abnormalities. Skeletal Radiol. 1980;5(4):233–9.
7. Xiang L, Luo M, Yan J, Liao L, Zhou W, Deng X, et al. Combined use of Clauss and prothrombin time-derived methods for determining fibrinogen concentrations: Screening for congenital dysfibrinogenemia. J Clin Lab Anal. 2018 May;32(4):e22322.
8. Simurda T, Asselta R, Zolkova J, Brunclikova M, Dobrotova M, Kolkova Z, et al. Congenital Afibrinogenemia and Hypofibrinogenemia: Laboratory and Genetic Testing in Rare Bleeding Disorders with Life-Threatening Clinical Manifestations and Challenging Management. Diagnostics. 2021 Nov 19;11(11):2140.
9. Bolton-Maggs PHB, Perry DJ, Chalmers EA, Parapia LA, Wilde JT, Williams MD, et al. The rare coagulation disorders--review with guidelines for management from the United Kingdom Haemophilia Centre Doctors’ Organisation. Haemophilia. 2004 Sep;10(5):593–628.
10. Szanto T, Lassila R, Lemponen M, Lehtinen E, Neerman-Arbez M, Casini A. Whole Blood Thromboelastometry by ROTEM and Thrombin Generation by Genesia According to the Genotype and Clinical Phenotype in Congenital Fibrinogen Disorders. Int J Mol Sci. 2021 Feb 25;22(5).
11. Casini A, de Moerloose P. Fibrinogen concentrates in hereditary fibrinogen disorders: Past, present and future. Haemophilia. 2020 Jan;26(1):25–32.