Rebelión mieloide
Myeloid rebellion
Del Castillo M, Figueroa F, Pagani M, Luciardi G, Zevallos C, Abdala A, Gómez R, Gramajo M.
Dpto. Hemato - Oncología y UTCPH, Hospital Néstor Kirchner. Tucumán. Argentina.
Palabras claves: neoplasias mieloproliferativas Filadelfia negativas,
LMC,
coexistencia.
Keywords: Philadelphia negative myeloproliferative neoplasms,
CML,
coexistence.
Todas las personas autoras han efectuado una contribución sustancial a la concepción o el diseño del estudio o a la recolección, análisis o interpretación de los datos, han participado en la redacción del artículo o en la revisión crítica de su contenido intelectual, han aprobado la versión final del manuscrito y son capaces de responder respecto de todos los aspectos del manuscrito de cara a asegurar que las cuestiones relacionadas con la veracidad o integridad de todos sus contenidos han sido adecuadamente investigadas y resueltas.
Resumen
Se presenta un paciente varón con diagnóstico inicial de una neoplasia mieloproliferativa Filadelfia negativa, sin respuesta al tratamiento y con empeoramiento de síntomas, por lo que se realiza nueva evaluación de enfermedad con diagnóstico de leucemia mieloide crónica coexistente. Evoluciona con anemia con requerimiento transfusional, suponiendo un reto diagnóstico y terapéutico para el equipo tratante, llevando a una nueva re evaluación del paciente y a la realización de consultas a expertos en el tema para la búsqueda de estrategias para su tratamiento. Se realiza una revisión bibliográfica del tema que se describe a continuación.
Abstract
We present a male patient with first diagnosis of Philadelphia chromosome negative myeloproliferative neoplasm (MPN), with no response to treatment and worsening of his symptoms. In further studies, stands out the coexisting diagnosis of chronic myeloid leukemia (CML) and afterwards the patient develops anemia requiring transfusions. The characteristics of this case represented a diagnostic and therapeutic challenge for the treating medical team and led to new re-assessments of the patient's situation, as well as consults with experts in the field in order to search for better strategies. A bibliographical review of the subject is described below.
Caso clínico
Paciente varón de 58 años de edad con antecedentes de hipertensión arterial, diabetes mellitus tipo 2 y colitis ulcerosa (CU), sin seguimiento adecuado de dichas patologías. Consulta en el año 2007, a sus 43 años de edad, por dolor abdominal. Se constata en laboratorio poliglobulia (Hto 67%, Hb 20 gr/dl), trombocitosis (plaquetas 1.273.000/mm3) y leucocitosis con neutrofilia (GB 14.000/mm3 con 78% neutrófilos segmentados). Al examen físico presenta esplenomegalia. No accede a realizarse estudio de medula ósea, por lo que se solicita mutación JAK2 V617F, siendo detectable. Se diagnostica policitemia vera (PV) de alto riesgo, iniciando tratamiento con hidroxiurea (HU), con mala adherencia (toma esporádica ante dolor abdominal). En 2015 consulta por primera vez a nuestra institución por exacerbación de dolor abdominal, que no cede con HU, asociado a pérdida de peso. Al examen físico presenta abdomen distendido con esplenomegalia a 8 cm por debajo del reborde costal (bazo de 200 mm por ecografía). Se realiza re evaluación de enfermedad presentando en estudios realizados: Hb 12,5 gr/dl, Hto 37%, GB 56.700/mm3 (segmentados 46%, basófilos 3%, eosinófilos 2%, linfocitos 6%, promielocitos 12%, mielocitos 9%, metamielocitos 9% y blastos 3%), plaquetas 939.000/mm3, estudio citogenético de médula ósea (MO) 46,XY, der(22)t(9;22)(q34;q11)[20], biopsia de MO con celularidad del 80%, con relación mielo-eritroide 4/1, trama de reticulina aumentada y mielofibrosis grado 1 (MF1) (Figura 1). Estudio molecular qPCR BCR-ABL1 p210 indetectable. No se realiza transcriptos atípicos (p190 - p230). Con los hallazgos antes mencionados se plantea el diagnóstico de leucemia mieloide crónica (LMC) Ph+, JAK2+ en fase crónica con Sokal de alto riesgo y Hasford de riesgo intermedio.
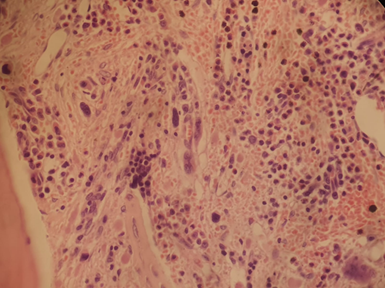
Figura 1. Anatomía patológica de MO con hallazgos compatibles con síndrome mieloproliferativo crónico con aumento de trama de reticulina (MF1).
Inicia inhibidores de tirosina kinasa (ITK) de 1ª generación (imatinib 400 mg/día) con buena tolerancia y adherencia. En el control al tercer mes de tratamiento persiste con leucocitosis, trombocitosis y elevación de LDH, es decir sin respuesta hematológica (RH), la que se interpreta secundaria a la mutación JAK2 detectable asociada. Al sexto mes alcanza respuesta citogenética completa (RCC) y BCR-ABL1 p210 no detectable. Al año de tratamiento presenta pérdida de RCC (20% Ph+) y BCR-ABL1 p210 no detectable (transcriptos p190 y p230 no fueron autorizados). Se interpreta como falla terapéutica. Por sus antecedentes personales se decide, como opción de tratamiento de 2° línea, dasatinib 100 mg/día (ITK de 2ª generación), iniciado en 2016 con buena tolerancia y adherencia. Alcanza RCC al tercer mes de iniciado el tratamiento y mantiene dicha respuesta en controles (mes 6, 12 y anuales) posteriores.
Evoluciona con complicaciones. En agosto del 2018 intercurre con reactivación de CU, por lo que inicia tratamiento con mezalasina con buena respuesta. Posteriormente, en octubre del 2020 presenta dolor abdominal leve con progresión de la esplenomegalia (bazo de 245 mm). Además, en control cardiológico semestral se detecta deterioro de la función sistólica y alteraciones de la motilidad parietal. Se realiza angioplastia de lesión severa en arteria circunfleja con reperfusión miocárdica, iniciando doble antiagregación. Al año siguiente, en el mes de mayo del año 2021 agrega en laboratorio anemia y disminución de trombocitosis habitual con aparición de blastos, eritroblastos (EB) y dacriocitos en sangre periférica (SP) con perfil férrico normal. Progresa en junio con requerimiento transfusional e incremento de EB y dacriocitos, iniciando eritropoyetina (EPO). Finalmente, en julio 2021 se realiza re evaluación de enfermedad persistiendo con RCC, qPCR BCR-ABL1 p190 y p210 negativos y con nueva anatomía patológica de MO que informa neoplasia mieloproliferativa (NMP) vinculable a mielofibrosis (MF) estadio fibrótico (Figuras 2-3-4).
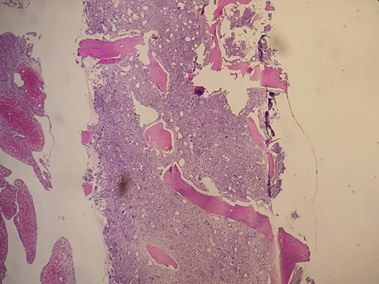
Figura 2. Biopsia de MO en 10x con celularidad poco evaluable por fibrosis. Se observa fibrosis con HyE probable MF3.
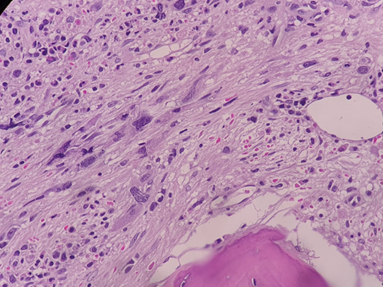
Figura 3. Biopsia de MO en 100x con arquitectura alterada por fibrosis estromal marcada. Relación mielo-eritroide: 3/1. Serie eritroide con disminución universal. Escasa maduración. Serie mieloide con elementos maduros y semimaduros prominentes. Aislados elementos inmaduros.
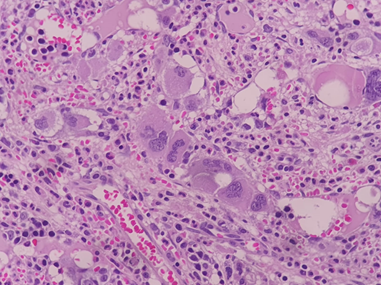
Figura 4. Biopsia de MO en 100x. En zonas de mayor celularidad se observa serie megacariocítica con aumento numérico relativo y absoluto, con marcados rasgos dismórficos y atipia celular con grupos alineados y distribución universal. Cambios estromales marcados con fibrosis, neovascularización, dilatación sinusoidal e histiocitos con hemosiderina. El tejido óseo presenta depósitos osteoides y aumento de actividad osteoblástica.
Luego de realizar la re evaluación del paciente se interpreta como LMC Ph+, JAK2+ con progresión a MF en estadio fibrótico. Se decide asociar ruxolitinib al tratamiento con dasatinib y EPO presentando buena tolerancia. En marzo de 2022 presenta leve disminución de esplenomegalia (bazo 238 mm por ecografía) y sin requerimiento transfusional.
Por último, en julio del año 2022 al no contar con hermanos histoidénticos, se deriva a centro de mayor complejidad para trasplante haploidéntico con su hijo, considerándose la necesidad de trasplante histoidéntico no relacionado. Se inicia la búsqueda internacional del donante.
Discusión
Las NMP son trastornos clonales caracterizados por la proliferación de una célula madre hematopoyética que produce la expansión de una o más series de linaje mieloide. Se dividen en NMP Filadelfia positivas (Ph+) y Filadelfias negativas (Ph-). Se propone que para el desarrollo de estas patologías se requiere la existencia de una inestabilidad genómica en la célula madre que conduce al desarrollo de lesiones moleculares que generan el fenotipo mieloproliferativo. La causa subyacente de esta inestabilidad no se comprende bien. La presencia del gen de fusión BCR-ABL1 conduce a un fenotipo de LMC, mientras que las mutaciones en los genes JAK2, CALR o MPL se vinculan a las NMP Ph-, representadas por la policitemia vera (PV), la trombocitemia esencial (TE) y la MF. Estas aberraciones genéticas dan como resultado tirosinas kinasas desreguladas que generan señales proliferativas en las células que dan inicio a la enfermedad. Dado que estas alteraciones generalmente son mutuamente excluyentes, una vez diagnosticada una NMP, rara vez se realizan determinaciones para las otras mutaciones. Sin embargo, hay varios informes de la existencia sincrónicas de LMC y MFP o PV. Las dos mutaciones líderes (Ph- y Ph+) pueden surgir de forma independiente o del mismo clon(1-3).
Se cree que la coexistencia de las NMP sones más frecuente de lo reportado debido al subdiagnóstico, por lo que representa un desafío terapéutico y además que, por los escasos datos, se desconoce su relevancia clínica(1,3).
Fisiopatología
En cuanto a la fisiopatología, se postulan dos teorías. La primera propone a la translocación BCR-ABL como un evento secundario en un clon mutado con JAK2, en tanto que la segunda plantea la presencia de mutaciones que surgen en clones independientes, dando lugar a un “tumor compuesto”. Ambos escenarios sugieren un genoma inestable que produce cambios en la célula hematopoyética y favorece la aparición de clones competidores(1,2,3,4).
Clasificación
Un análisis retrospectivo de casos de MD Anderson Cancer Center, donde se evaluó a pacientes con diagnóstico de NMP entre los años 2000 a 2017, de un total de 2274 pacientes, 16 presentaron dos mutaciones líderes coexistentes. Ellos proponen una clasificación interesante en dos categorías principales:
1. Pacientes con mutaciones líderes que incluyan al BCR/ABL1 + JAK2/MPL/CALR.
2. Pacientes con dobles mutaciones líderes en NMP Ph- pero negativos para BCR/ABL1(1).
Dentro de la categoría de pacientes con mutaciones líderes que incluyan al BCR/ABL1 + JAK2/MPL/CALR, a su vez existen dos subgrupos. El primero representado por pacientes con la presencia de transcripto BCR-ABL1 clínicamente no relevante, detectado incidentalmente y NMP Ph− coexistentes, que se caracterizan por la detección de niveles bajos de BCR/ABL1 en personas sanas, lo que se define como hematopoyesis clonal de potencial indeterminado (CHIP), condición que tiende a incrementarse con la edad. Sin embargo, para el desarrollo de la enfermedad se deben adquirir cambios adicionales, sumado a que estas anomalías suelen desaparecer con el tiempo, confirmando que representan un hallazgo incidental sin significancia patológica(1).
El segundo subgrupo se refiere a pacientes con mutaciones líderes clínicamente relevantes: LMC Ph+ y NMP Ph- coexistentes. Se destaca en dichas situaciones las evoluciones diversas: rápida progresión de la LMC a crisis blástica, remisión de ambas NMP con trasplante de células progenitoras hematopoyéticas (TCPH) o enfermedad estable con respuesta molecular de la LMC. Aquí la importancia de la anatomía patológica de MO para alertar sobre la posibilidad de NMP concomitantes. Es fundamental prestar atención a las características de los megacariocitos. Por ejemplo, megacariocitos grandes y atípicos con o sin mielofibrosis, en una médula con hallazgos compatibles con una LMC darían indicios para solicitar la búsqueda de la mutación BCR-ABL1 y además mutaciones en JAK2, CALR y MPL(1,3).
La coexistencia de mutaciones líderes JAK2/MPL/CALR puede tener implicancias en la respuesta a los inhibidores de tirosina kinasa (ITK) en el tratamiento de la LMC, en algunos casos con peores resultados. El tratamiento dirigido hacia un blanco molecular produce una presión selectiva sobre los clones de la enfermedad, por lo que se sugiere un tratamiento combinado para alcanzar mejores respuestas, inclusive teniendo en cuenta el riesgo de la toxicidad(1,3,4).
Por último, se encuentra la segunda categoría de pacientes con dobles mutaciones líderes en NMP Ph- No BCR/ABL1. Existe un subregistro de la verdadera frecuencia de esta entidad, por el hecho de que la determinación de las tres mutaciones no se realiza de rutina en todos los pacientes, por lo que se desconoce la verdadera relevancia de la presencia de mutaciones líderes coexistentes(1).
Elección de los ITK
Para la elección de los ITK en pacientes con mutaciones líderes coexistentes, la fibrosis debe ser considerada como un parámetro anatomopatológico importante para la decisión del tratamiento inicial, siendo además un marcador de pronóstico independiente. Existiría un beneficio de los ITK de segunda generación (4-7).
Otros autores refieren falla al tratamiento con los ITK en pacientes JAK2 y NMP Ph+, dado por falta de aclaramiento del transcripto BCR-ABL1, que llevaría a la progresión de la enfermedad. Esta situación se reproduce en casos de pacientes con NMP CALR/MPL+ y NMP Ph+, no estando asociado a toxicidad o mala adherencia al tratamiento. Faltan estudios para comprender y certificar esta situación (1,3,4).
Mielofibrosis secundaria y tratamiento con ruxolitinib
La MF es una NMP crónica caracterizada por la expansión clonal de una célula madre pluripotente. Puede ser primaria o secundaria. La sintomatología es variada, desde astenia y adinamia hasta hematopoyesis extramedular y, como consecuencia, la esplenomegalia en un 90% de los pacientes (7).
Existen múltiples escalas pronósticas vigentes para categorizar a los pacientes con MF. El IPSS fue diseñado para ser aplicado al diagnóstico, mientras que el DIPSS y DIPSS plus permiten predecir sobrevida en cualquier momento de la evolución de la enfermedad. El índice MYSEC fue específicamente diseñado para MF secundaria(7,8).
Para la elección del tratamiento se tiene en cuenta la gran variabilidad clínica de la MF. El ruxolitinib es un inhibidor potente y selectivo de las quinasas asociadas a Janus (JAK) JAK1 y JAK2. No existen diferencias en las tasas de respuesta en pacientes JAK2 positivos o negativos, por lo que su indicación es independiente del estado mutacional. Por lo tanto, este fármaco es de elección en pacientes con MF primaria o post PV/TE con esplenomegalia sintomática y/o síntomas constitucionales. El inicio del tratamiento en etapas más tempranas permite utilizar mayores dosis y una mejor tolerancia(7).
Rol del TCPH en mielofibrosis secundaria
Si bien el número de trasplantes en MF aumentó constantemente, la disponibilidad de tratamientos médicos efectivos plantea la necesidad de una mejor evaluación y selección de los pacientes a trasplantar. Los índices clásicos IPSS, DIPSS y DIPSS plus son malos predictores para los pacientes con MF secundaria. Por esta razón se utiliza el índice pronóstico MYSEC, siendo candidatos a trasplante aquellos pacientes con un MYSEC intermedio 2 o alto(8).
El uso de ruxolitinib previo al TCPH acelera el injerto y mejora su función, además de disminuir la incidencia y la gravedad de la enfermedad de injerto contra huésped (EICH) y la mortalidad sin recaídas, y por estas razones se indica previo al TCPH. Por otro lado, la esplenectomía pre TCPH para pacientes con MF sigue siendo un tema de controversia y hasta la fecha se han reportado resultados contradictorios. El procedimiento parece acelerar la recuperación hematológica posterior al TCPH en función de un injerto más rápido, pero su impacto en la tasa de recaídas y la supervivencia no es aún claro (9-11).
Conclusiones
Cada vez existe más evidencia de la presencia de NMPs co-existentes. Existen dos teorías que explican la fisiopatología de esta entidad. Por un lado, la presencia de dos clones distintos, y por otro la existencia de dos mutaciones en el mismo clon. Ambos escenarios actuarían sobre un genoma inestable.
Se debe considerar el diagnóstico de una NMP Ph- coexistente cuando existen discrepancias entre la respuesta molecular/citogenética y la respuesta clínica en los pacientes con LMC y viceversa.
El diagnóstico de estas patologías presentes en forma simultánea continúa siendo un desafío diagnóstico y terapéutico, reafirmando la importancia del trabajo interdisciplinario y que permita ofrecer un tratamiento combinado.
Bibliografía
1. Boddu P, Chihara D, Masarova L y col. The co-occurrence of driver mutations in chronic myeloproliferative neoplasms. Ann Hematol;. 2018;97(11):2071-2080.
2. Kandarpa M, Wi Y, Robinson D, Burke P, Chinnaiyan A, Talpaz M. Clinical characteristics and whole exome/transcriptome sequencing of coexisting chronic myeloid leukemia and myelofibrosis. Am J Hematol. 2017;92:555-561.
3. Montivero A. JAK 2 y BCR-ABL. ¿Son siempre mutuamente excluyentes? Hematología. 2021;25:147-150.
4. Yi JH, Kim HR. Coexistence of BCR/ABL1-positive chronic myeloid leukemia and JAK2 V617F-mutated myelofibrosis successfully treated with dasatinib and ruxolitinib. Blood Res. 2019;54(1): 77-79.
5. Hussein K, Bock O, Seegers A et al. Myelofibrosis evolving during imatinib treatment of a chronic myeloproliferative disease with coexisting BCR-ABL translocation and JAK2V617F mutation. Blood. 2007;109:4106-4107.
6. Eliacik E, Isik A, Aydin C y col. Bone marrow fibrosis may be an effective independent predictor of the 'TKI drug response level' in chronic myeloid leukemia. Hematology. 2015;20(7):392-6.
7. Tefferi A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am J Hematol. 2021;96:145-162.
8. Polverelli N, Farina M, D’Adda M y col. How We Manage Myelofibrosis Candidates for Allogeneic Stem Cell Transplantation. Cells. 2022;11(3),553.
9. Salit R, Scott B, Stevens E y col. Pre-hematopoietic cell transplant Ruxolitinib in patients with primary and secondary myelofibrosis. Bone Marrow Transplant. 2020; 5:70-76.
10. Robin M, Zine M, Chevret S y col. The impact of splenectomy in myelofibrosis patients before allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2017;23:958-964.
11. Helbig G, Wieczorkiewicz‑Kabut A, Markiewicz M y col. Splenic irradiation before allogeneic stem cell transplantation for myelofibrosis. Med Oncol. 2019 Jan 8;36(2):16.