Linfoma del manto y linfoma difuso de células grandes B: un paciente, dos entidades
Mantle lymphoma and diffuse large B - cell lymphoma: one patient, two entities
Olsen, S; Basso, L; Siles, R.; Gutiérrez, R.; García, C
Hospital San Bernardo, Salta - Argentina
Palabras claves: linfoma,
células,
manto,
difuso B,
histología.
Keywords: lymphoma,
cells,
mantle,
diffuse B-cells,
histology.
Resumen
El linfoma de células del manto (LCM) se origina en los linfocitos B pre-germinales de la zona interna del manto del folículo linfoide. Dentro de las características inmunofenotípicas, la marcación nuclear con CCD-1 (ciclina D1) es expresada en más de 95% de los casos(1). Se presenta el caso clínico de un paciente de 56 años con diagnóstico de LCM al cual se le realizó quimioterapia sistémica alcanzando remisión completa y que después de siete meses de seguimiento evoluciona con recaída temprana, se interviene con nueva toma de biopsia y estudios de inmunohistoquímica, realizando diagnóstico de linfoma difuso de células grandes B (LDCGB). Ante esta situación nos planteamos si un LCM pudo evolucionar con LDCGB o si el diagnóstico inicial de LCM pudo ser confundido con un LDCGB. En este artículo, nos proponemos informar sobre la presentación clínica e histopatológica de este caso con una revisión de la literatura.
Abstract
Mantle cell lymphoma (MCL) originates from the pre-germinal B cells of the inner mantle zone of the lymphoid follicle. As an immunophenotypic characteristic, nuclear CCD-1 (cyclin D1) labeling is expressed in over 95% of the cases(1) . We report a 56-year-old patient diagnosed with MCL who underwent systemic chemotherapy with complete remission and early relapse after a 7- month follow-up. The patient underwent a new biopsy and immunohistochemistry investigations, and a diagnosis of diffuse large B-cell lymphoma (DLBCL) was established. Given this scenario, the question was raised as whether an MCL progressed with DLBCL or whether the initial MCL diagnosis was confused with a DLBCL. Herein we aim to report on the clinical and histopathological manifestations of this case based on a literature review.
Introducción
De todos los linfomas no Hodgkin (LNH), el LCM representa 6-9%, se origina en los linfocitos B (naïve pre-germinales) de la zona interna del manto del folículo linfoide(1). El componente más importante en la patogénesis está determinado por una disrupción simultánea de los mecanismos implicados en la regulación del ciclo celular con sobreexpresión de ciclina D1(2). En cuanto a la histopatología, esta neoplasia puede presentar un patrón de crecimiento variable en tres tipos: área del manto, nodular o difuso; las células son pequeñas a medianas, monomorfas, de núcleos con irregularidad leve a marcada, cromatina dispersa y nucléolo poco conspicuo. Un 20% de los casos puede mostrar una citología que remeda linfoblastos, con células de tamaño mediano, “variante blastoide” y con menor frecuencia se pueden ver “variantes pleomórficas” o de células pequeñas que remedan linfoma linfocítico o linfoma de la zona marginal. Estas dos últimas variantes de curso agresivo y peor pronóstico. En cuanto a las características inmunofenotípicas con técnicas de inmunohistoquímica expresan CD20+, CD5+, CD43+, BCL2+, siendo negativos para CD23, CD10, bcl-6 y MUM-1. La marcación nuclear con CCD-1 es expresada en más de 95% de los casos. Existen casos raros negativos para la expresión de CCD-1 que expresan CCD-2 o CCD-3 y casos con inmunofenotipo aberrante como ausencia de CD5. El índice de proliferación mayor de un 30% demostrado con MIB-1 (Ki67) se asocia a un peor pronóstico. El diagnóstico diferencial debe realizarse según el patrón de crecimiento, en las variantes pleomórficas puede confundirse con LDCGB(1). Esta entidad se caracteriza por representar un 35% de todos los LNH, pueden haberse originado de novo o bien secundario a un linfoma folicular, de la zona marginal y de Hodgkin variante predominio linfocítico nodular. En cuanto a la histopatología, las células tienen aspecto morfológico variable, generalmente son células linfoides grandes con infiltración uniforme que crecen en forma difusa, contienen abundantes mitosis y áreas de necrosis, que en algunos casos pueden ser muy extensas y entorpecer el diagnóstico. Se clasifican según la variante morfológica, (centroblástico, inmunoblástico, anaplásico, entre otros) en subtipos moleculares, (centro germinal o no centro germinal) y en entidades distintivas. El inmunofenotipo expresa marcadores B: CD20, CD79a y PAX-5. El algoritmo de Hans utiliza tres marcadores para diferenciar entre los subtipos moleculares: el CD10, BCL-6 y IRF4/MUM-1. La proteína bcl-2 es positiva en un 50% de las células tumorales. El índice de proliferación expresado por MIB-1 (Ki67) generalmente es mayor del 40%, cuando supera el 90 % se trata de variantes altamente proliferativas. Un 5 a 10 % de los casos expresa CD5, los cuales se distinguen del LCM variante blastoide por la ausencia de CCD-1(3). Sin embargo, se han reportado casos extremadamente raros en la que esta neoplasia puede albergar reordenamientos de CCND1, con expresión de la proteína ciclina D1(2), en estos casos SOX-11 es una técnica de inmunohistoquímica útil para diferenciarlos, ya que se encuentra negativo para los LDCGB y positivo en más del 90% de los casos de LCM incluyendo los CCD-1 negativos(4). A continuación se presenta un caso clínico en el que se detalla la presentación de un paciente con diagnóstico de LCM que presenta como recaída temprana progresión a LDCGB.
Caso clínico
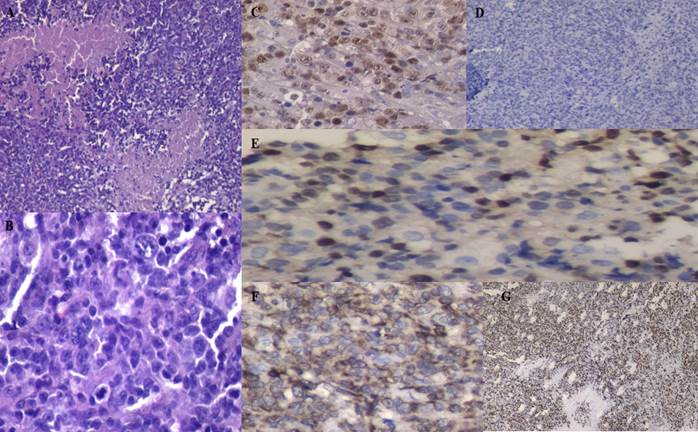
Paciente de sexo masculino, de 56 años de edad, proveniente de la ciudad de Salta sin cobertura social, refiere como antecedentes tóxicos ser estilista ocasional (CAGE: 2) y ex tabaquista severo con abandono hace diez años. Inició con poliadenopatías palpables, sudoración nocturna y pérdida de peso en noviembre de 2019, por lo que consultó un mes más tarde en hospital de origen. Fue valorado por servicio de Clínica Médica que solicitó tomografía computada (TC) de tórax, abdomen y pelvis evidenciándose en cuello adenopatías en triángulos posteriores, en mediastino pre vascular, pre traqueal, ventana aorto-pulmonar e infracarinal, así como también en ambas regiones axilares, retroperitoneales, intero-cavo aórticas y latero aórtica, con múltiples conglomerados adenomegálicos en raíz del mesenterio y en región inguinal. Se le realizó biopsia incisional de adenopatía cervical con resultado anatomopatológico que informó presencia de un parénquima ganglionar con proliferación difusa de células mononucleares de mediano tamaño con macrocariosis de bordes irregulares, con cromatina densa y núcleos prominentes, en general regular en cuanto tamaño y forma, se reconocieron frecuentes figuras de mitosis atípicas. Se recibió inmunohistoquímica (Figura 1) realizando diagnóstico de LCM.
El paciente acude a consulta en el Servicio de Hematología en marzo del 2020 por progresión de síntomas. Al examen físico se presenta con hábito asténico, adelgazado, ECOG 2, lúcido con suficiencia cardio-respiratoria y poliadenopatías palpables. Asiste a primera valoración con estudios realizados y resultados de laboratorio; glóbulos blancos: 16.170/mL (42/2/0/49/7), hemoglobina: 10.0 g/dL, hematocrito: 33%, plaquetas: 337.000/mL, láctico deshidrogenasa: 513 U/L con parámetros de lisis tumoral alterados y b2 microglobulina: 5.4 U/ml. Se le realiza punción aspiración de médula ósea con obtención de muestra para citogenético que informo cariotipo conservado XY en 21 metafases analizadas, como así también se efectúa biopsia que descarta compromiso medular. Se le solicita video-endoscopía digestiva alta (VEDA) en la que se toman muestras y cuyo resultado anatomo patológico informa compromiso gastrointestinal por igual entidad linfoproliferativa.
Ante un paciente joven, sin comorbilidades significativas, con diagnóstico de LCM estadío III B, MIPI riesgo alto, con deterioro clínico por la progresión de la enfermedad, nos planteamos como equipo tratante realizar esquema de inducción con 3 R-CHOP/ 3 R-DHAP (alternados) y consolidar con trasplante autólogo de médula ósea (TAMO), por lo que se inicia solicitud de derivación. El paciente inicia tratamiento en abril del 2020 con buena tolerancia al mismo. Como complicaciones presenta citopenias acentuadas luego de la infusión de R-DHAP, en una oportunidad con requerimiento de colocación de estimulante de colonias granulocíticas, anemia y trombocitopenia sin requerimiento transfusional; además presenta una trombosis venosa extensa en miembro inferior izquierdo luego del segundo ciclo de quimioterapia, por lo que realiza anticoagulación con enoxaparina. Se finaliza sexto ciclo en noviembre de 2020, un mes más tarde se realiza TC de control en la que constata resolución de poliadenopatías y VEDA que informa gastropatía eritematosa y ulcera gástrica sin compromiso de enfermedad por histología.
En enero de 2021, ante la falta de posibilidad de derivación para TAMO, se decide realizar un ciclo adicional de R-CHOP/RDHAP. Se asume al paciente en remisión completa.
El paciente progresa en marzo de 2021, con dolor e impotencia funcional de miembro inferior izquierdo. Es valorado por Traumatología con diagnóstico de fractura patológica supracondílea en fémur izquierdo por lo que se realiza tratamiento ortopédico en primera instancia. Por resonancia magnética nuclear (RMN) se evidencia imagen blástica expansiva con deformación de la estructura ósea y afectación articular, por lo que se solicita realizar biopsia en instancia de colocar la prótesis correspondiente. En julio de 2021 acude a control en Servicio de Hematología presentando al examen físico ganglio palpable inguinal izquierdo que se decide biopsiar. Se recibe informe anatomopatológico que describe fragmentos disgregados del parénquima ganglionar con remplazo de la arquitectura por una proliferación neoplásica constituida por elementos linfoides de tamaño grande con nucléolos prominentes, mitosis abundantes y áreas extensas de necrosis; por lo que se solicita inmohistoquímica (Figura 2).
Ante el diagnóstico de LDCGB, se presenta el caso clínico en Comité de Tumores intrahospitalario, se realiza ateneo multidisciplinario y se discute la posibilidad de una recaída temprana con otra entidad linfoproliferativa. Se concluye solicitar CCD-1 que se informa como negativo en segunda instancia y re estadificación del paciente.
En agosto de 2021 el paciente ingresa al nosocomio por dolor total con intervención de Cuidados Paliativos; durante internación intercurre con neumonía intrahospitalaria que conduce al deceso.
Discusión
Se presenta un caso clínico de un paciente joven, sin comorbilidades significativas, con diagnóstico de LCM en estadío avanzado, que realizó quimioterapia intensiva con buena tolerancia alcanzando objetivo terapéutico y que durante seguimiento presenta recaída temprana con diferente entidad linfoproliferativa.
Ante esta situación nos planteamos si el diagnóstico inicial de LCM pudo ser confundido con un LDCGB. Las variantes pleomórficas del LCM se presentan con células de tamaño intermedio-grande, núcleos irregulares con cromatina dispersa, a veces nucléolo pequeño e índice mitótico elevado. Estas características concuerdan con la presentación en nuestro paciente, en este caso la inmuhistoquímica que coincidió con el inmunofenotipo nos condujo al diagnóstico de LCM.
Sin embargo, se han reportado casos extremadamente raros en los que el LDCGB puede albergar reordenamientos de CCND1, con expresión de la proteína CCD-1(2). En estos casos SOX-11 sería una técnica de inmunohistoquímica útil para diferenciarlos, ya que se encuentra negativo para los LDCGB y positivo en más del 90% de los casos de LCM incluyendo los CCD-1 negativos(4). Esta técnica nos ayudaría a dilucidar si un LCM pudo recaer con LDCGB o si inicialmente nos encontrábamos con un diagnóstico que nos llevó a confundir la entidad.
En agradecimiento a la Dra. Gonorasky, S.
Bibliografía
1. Korin L, Agriello E, Alfonso G y col. Linfoma del Manto. Guías de Diagnostico y Tratamiento. Sociedad Argentina de Hematología 2021; 29-36.
2. Andrew M, Parrott AM, Haggiagi V y col. Linfoma primario de células B grandes del sistema nervioso central con expresión de ciclina D1 y t(11;14) ( IGH- CCND1): ¿linfoma difuso de células B grandes con reordenamiento de CCND1 o linfoma de células del manto? Hematological Oncology. 2020;38:817-822.
3. Korin L, Agriello E, Alfonso G y col. Linfoma Difuso de Células Grandes B. Guías de Diagnóstico y Tratamiento. Sociedad Argentina de Hematología 2021; 5-21.
4. Hsiao SC, Cortada IR, Colomo L y col. SOX11 es útil para diferenciar el linfoma difuso de células B grandes positivo para ciclina D1 del linfoma de células del manto. Histopathology. 2012;61:685-693.
5. Wen YC, Chang GJ, Chang TH y col. Linfoma pleomórfico de células del manto que imita morfológicamente al linfoma difuso de células B grandes: negatividad común para ciclina D1 y un algoritmo inmunohistoquímico simple para evitar el escollo diagnóstico. Hystopathology. 2017;70:986-999.
Figura 1
|
|
|
Figura 1. Fotografías de los preparados de adenopatía cervical. A: tinción hematoxilina eosina, B: CD 45 positivo, C: CD 20 positivo, D: PAX 5 positivo nuclear, E: BCL 2 positivo, F: CCD-1 positivo, G: KI67 positivo con índice de proliferación del 70%, H: CD 10 positivo, I: CD 3 negativo con T remanentes. |
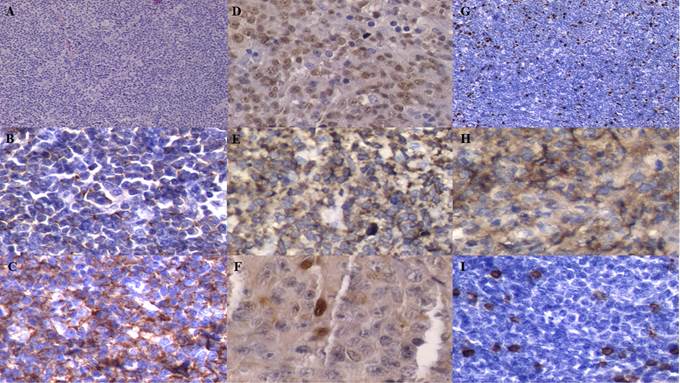
Figura 2
|
|
|
Figura 2. Fotografía de los preparados de adenopatía inguinal. A: tinción con hematoxilina eosina con extensas áreas de necrosis. B: tinción con hematoxilina eosina en la que se visualiza parénquima ganglionar con células grandes. C: PAX 5 positivo, D: CD 30 negativo, E: MUM 1 patrón nuclear positivo; F: BCL 2 positivo, G: KI 67 con índice de proliferación del 75%. |