Linfohistiocitosis hemofagocítica (HLH) en pacientes pediátricos. Experiencia de una institución
Hemophagocytic lymphohistiocytosis (HLH) in pediatric patients. Experience of an institution
Deana A1; Sanchez Tovar E1; Sossi ME1; Segovia Gira CV1; Morici M1; Merhar C2; Riccheri C1
1. Servicio de Hematología y Oncología Pediátrica. Hospital Nacional Dr. A. Posadas. Bs As. Argentina
2. Sección de Inmunología Pediátrica. Hospital Nacional Dr. A. Posadas. Bs As. Argentina
email: eduardosancheztovarmd@gmail.com
Palabras claves:
Keywords:
Resumen
La linfohistiocitosis hemofagocítica (HLH) es una entidad clínica poco frecuente, potencialmente mortal. Este síndrome describe a pacientes con hiperinflamación sistémica grave. El diagnóstico se realiza en función de criterios clínicos, genéticos y de laboratorio. Se describen dos tipos principales de HLH: primaria y secundaria. La HLH primaria está asociada a defectos genéticos en: perforina, UNC13D, sintaxina 11, sintaxina BP2, moléculas intervinientes en el mecanismo de citotoxicidad celular. Las HLH secundaria (sLHL) se asocian a causas infecciosas (virus Epstein Barr -VEB-, tuberculosis -TBC-, brucelosis, leishmaniasis, SARS-CoV-2), reumatológicas (síndrome de activación macrofágica) y hematooncológicas.
Material y métodos. Realizamos un estudio retrospectivo, descriptivo, observacional entre enero de 2012 hasta octubre de 2022. Se analizaron los datos clínicos y de laboratorio de 12 pacientes (ptes) con HLH atendidos en nuestro centro.
La mediana de edad fue 7,25 años (r: 1,1 a 14,9), 6 varones y 6 mujeres. Los 12 ptes fueron sHLH. Manifestaciones clínicas: fiebre persistente en todos los pacientes, hepatomegalia (58%), esplenomegalia (92%) y síntomas del sistema nervioso central (16%). 8 ptes (67%) tenían infección por VEB. En 6 ptes (50%) se realizó estudio de biología molecular. 11 ptes recibieron tratamiento de acuerdo al protocolo HLH-2004 con fase de inducción a la remisión de 8 semanas que incluye: dexametasona, etopósido, ciclosporina, con/sin metotrexate intratecal semanal, seguida de una fase de mantenimiento de hasta 40 semanas con pulsos de dexametasona, etopósido y ciclosporina. 1 pte recibió tratamiento de la enfermedad de base más ciclosporina.
Resultados. La tasa de mortalidad al año fue de 25%. Tras una mediana de seguimiento de 15 meses (rango: 10 días - 129 meses), 3 ptes fallecieron, 9 ptes sobrevivieron. La sobrevida global (SG) fue 75%.
Conclusiones. La HLH se caracteriza por la activación patológica del sistema inmune. No existe ninguna medida preventiva conocida hasta el momento. Se define gracias a una serie de criterios diagnósticos establecidos por la Sociedad de Histiocitosis. De cumplirse los criterios diagnósticos debe instaurarse precozmente el tratamiento, dado que esta patología presenta una alta tasa de mortalidad.
En pacientes con formas primarias y alteración genética subyacente conocida, está indicado el consejo genético y el trasplante de células progenitoras hematopoyéticas como tratamiento curativo.
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a rare, life-threatening and generally underdiagnosed clinical entity. This syndrome describes patients with severe systemic hyperinflammation. The diagnosis is made on the basis of clinical, genetic and analytical criteria. Two main types of HLH are described: primary and secondary. Primary HLH is associated with genetic defects in: perforin, UNC13D, syntaxin 11, syntaxin BP2, molecules involved in the mechanism of cellular cytotoxicity. The sHLH are associated with infectious causes such as Epstein Barr virus (EBV), tuberculosis, brucellosis, leishmaniasis and SARS-CoV-2, rheumatologic (in this case it is defined as macrophage activation syndrome) and hemato-oncological causes.
Material and methods. We conducted a retrospective, descriptive, observational study between January 2012 and October 2022. The clinical and laboratory data of 12 patients (ptes) with HLH treated at our center were analyzed.
The median age was 7.25 years (r: 1.1 to 14.9). 6 men and 6 women. All 12 ptes were sHLH. Clinical manifestations: persistent fever in all patients, hepatomegaly (58%), splenomegaly (92%) and central nervous system symptoms (16%). 8 ptes (67%) had EBV infection. In 6 ptes (50%) a molecular biology study was carried out. 11 ptes received treatment according to the HLH-2004 protocol with an 8-week induction phase that included etoposide, dexamethasone, cyclosporine with/without weekly intrathecal methotrexate, followed by a maintenance phase of up to 40 weeks with dexamethasone pulses, VP16 and cyclosporine and 1 pte received treatment for their underlying disease plus cyclosporine.
Results. The 1-year mortality rate was 25%. After a median follow-up of 15 months (range: 10 days- 129 months), 3 ptes died, 9 ptes survived. Overall survival (OS) was 75%.
Conclusions. HLH is characterized by pathological activation of the immune system. There is no known preventive measure so far. It is defined by a set of diagnostic criteria established by the Histiocytosis Society. If the diagnostic criteria are met, treatment should be instituted early, since this pathology has a high mortality rate.
In patients with primary forms and known underlying genetic alteration, genetic counseling and hematopoietic progenitor cell transplantation are indicated as curative treatment.
Introducción
La linfohistiocitosis hemofagocítica (HLH) o síndrome hemofagocítico (SHF) es un cuadro hiperinflamatorio sistémico derivado de la desregulación del sistema inmune, en el que la activación permanente de células T citotóxicas, NK y macrófagos tisulares desarrollan el papel principal, que pone en riesgo la vida.
Scott y Robb-Smith describieron el síndrome por primera vez en 1939, como reticulosis histiocítica medular, en un paciente que presentaba un cuadro clínico de fiebre, adenopatías, pancitopenia y hemofagocitos en medula ósea(1).
En 1952 Farquhar y Claireux describieron la presentación pediátrica del cuadro como un desorden de la inmunidad(2).
En 1985 Claudio Griscelli y colaboradores utilizaron el término de “síndrome de activación macrofágico” (SAM) para describir siete pacientes con artritis idiopática juvenil (AIJ) que desarrollaron encefalopatía de inicio agudo, coagulopatía y hepatitis, con activación de macrófagos en las biopsias hepáticas(3).
De acuerdo a la bibliografía se describen dos tipos principales de HLH: primaria y secundaria.
La forma primaria o familiar está asociada a alteraciones inmunológicas secundarias a mutaciones genéticas. Por el contrario, cuando son desencadenadas por factores externos se constituye la forma secundaria (sHLH) o adquirida, en su mayoría causados por infecciones, tumores malignos y enfermedades autoinmunes(4-5).
En la HLH primaria se conoce la causa genética subyacente en aproximadamente un 30-70%. Pertenecen a este grupo las HLH familiares (FHLH). Hasta el momento se han descrito 5 formas familiares (FHLH 1-5) que se caracterizan por defectos en la vía de citotoxicidad de las células T y NK. En la tipo 1 aún no se conoce el defecto molecular, y de la 2 a la 5 se definen por variantes en los genes PRF1, UNC13D, STX11 y STXBP2. La FHLH tiene una incidencia estimada de alrededor de 1:50000 niños nacidos vivos(5-6). Es una enfermedad fatal con una mediana de supervivencia de menos de 2 meses después del diagnóstico si no se trata. Se manifiesta en los primeros años de vida, 70 a 80% dentro del primer año, y existe predisposición a manifestar episodios de HLH recurrentes. El compromiso de sistema nervioso central (SNC) es frecuente(7).
A pesar de su nombre, los antecedentes familiares suelen ser negativos, ya que la enfermedad es recesiva. Es importante destacar que la aparición de FHLH y los episodios de la enfermedad pueden ser provocados por infecciones(7).
Del mismo modo, pertenecen a las formas primarias inmunodeficiencias como: síndrome de Griscelli tipo 2, Chediak-Higashi, Hermansky-Pudlak tipo 2 o síndrome linfoproliferativo ligado al cromosoma X (XLP)(8-9).
La sHLH es más frecuente que las formas primarias, se han descripto como: complicación de inmunodeficiencias primarias, errores innatos del metabolismo, infecciones (generalmente asociadas a VEB), fúngicas, bacterias, parásitos (en particular leishmaniasis) y en los últimos dos años asociadas a SARS-CoV-2(10), procesos neoplásicos (especialmente hematológicos, aunque no exclusivos), así como de origen autoinmune.
Es difícil distinguir la HLH primaria y secundaria sobre la base clínica. Los hallazgos más típicos son fiebre, hepato-esplenomegalia y citopenias. Otros hallazgos comunes incluyen hipertrigliceridemia, coagulopatía con hipofibrinogenemia, disfunción hepática, niveles elevados de ferritina, transaminasas séricas y síntomas neurológicos que pueden estar asociados con hiperproteinemia del líquido cefalorraquídeo y pleocitosis moderada. Otros hallazgos clínicos pueden ser linfadenopatía, erupción cutánea, ictericia y edema(11-13).
El diagnóstico de la HLH es un diagnóstico genético, clínico y analítico. En la actualidad se realiza según los criterios del protocolo HLH-2004(11).
La alta tasa de mortalidad hace que el reconocimiento y el tratamiento oportuno de este síndrome hiperinflamatorio sean esenciales. Es importante conocer su existencia de manera precoz, por su rápida capacidad de generar afectación multiorgánica, y la instauración de un tratamiento eficaz en una fase temprana condiciona el pronóstico del paciente.
A continuación presentaremos el análisis retrospectivo de las HLH en el Servicio de Hemato-Oncología Infantil del Hospital Nacional Dr. A. Posadas.
Material y métodos
Se realizó un estudio descriptivo, retrospectivo, observacional en el período comprendido entre enero de 2012 a octubre de 2022. Se identificaron 12 ptes, de 1,1 años a 14,9 años. Todos los ptes cumplieron al menos cinco de los ocho HLH-2004 criterios diagnósticos(10) (Tabla1).
Las variables continuas se expresaron como medianas y rangos, mientras que las variables categóricas se expresaron como frecuencias y porcentajes. El análisis univariado se realizó con T-test para variables continuas y con χ2 o test de Fisher para variables categóricas. La supervivencia global (SG) se analizó mediante el método de Kaplan-Meier. El análisis estadístico se realizó con el programa SPSS 22.0.
Resultados
Ingresaron 12 ptes entre enero de 2012 hasta octubre de 2022. 6 varones y 6 mujeres (relación V:M 1:1); con una mediana de edad de 7,25 años (r: 1,1 a 14,9). La mediana de tiempo de diagnóstico fue 20,5 días (r: 10 a 40). Los 12 ptes fueron sHLH. 8 ptes (67%) presentaron infección por VEB (Tabla 2).
Todos los ptes presentaron fiebre al momento del diagnóstico. Los síntomas clínicos: hepatomegalia (58%), esplenomegalia (92%) y síntomas del SNC (16%). Las anormalidades de laboratorio más comunes fueron neutropenia (50%), anemia (92%), trombocitopenia (75%), hipofibrinogenemia (58%), hipertrigliceridemia (67%), hiperferritinemia (83%) y hemofagocitosis en MO (75%). Alrededor del 67% de nuestros pacientes tenían alteración en la función hepática (elevación transaminasas y bilirrubina), el 92% tenía LDH elevada y el 42% tenía hipoalbuminemia. Casi el 25% de los pacientes mostró daño en la función renal (Tabla 3).
En 6 ptes (50%) se realizó estudio de biología molecular y no se hallaron variantes patogénicas (Tabla 4).
11 ptes (92 %) recibieron el régimen HLH-2004 como tratamiento inicial (Gráfico 1),
1 pte (8%) recibió quimioterapia específicas de la enfermedad de base (LLA-B) más ciclosporina.
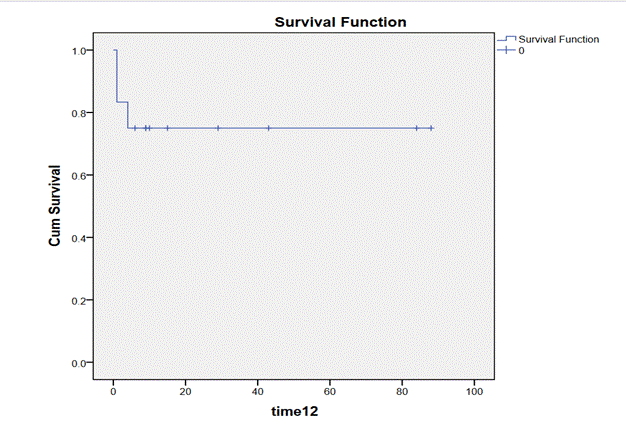
La tasa de mortalidad al año fue de 25%. Tras una mediana de seguimiento de 15 meses (rango: 10 días - 129 meses) 3 ptes fallecieron y 9 ptes sobrevivieron. La SG a 5.5 años de todos los pacientes fue del 75% (Gráfico 2).
Tabla 1. Criterios diagnósticos propuestos en HLH-2004

Tabla 3. Los parámetros clínicos, laboratorio y forma de presentación de los pacientes con HLH
|
Nº |
Hb/PMN/ Plq (g/dL, 103 x mm3) |
BiT/TGO TGP/LDH (UI/L) |
Albúmina (g/dL) |
Triglic (mg/dL) |
TP/KPT/ Fibrinóg (g/L) |
Ferritina (ng/ml) |
Urea/ Creat (mg/dl) |
Hemof MO |
Infección asociada |
Alter. cromos |
Forma presentación |
|
1 |
12.4/560/ 177 |
0.6/24/ 25/720 |
3.8 |
137 |
15/42/ 110 |
122 |
0,34/ 0,8 |
SI |
- |
NO |
NO |
|
2 |
7.8/840/ 82 |
4,5/162/84/612 |
4.1 |
281 |
12/30/ 295 |
721 |
1,9/3.3 |
SI |
- |
NO |
NO |
|
3 |
9/600/89 |
4.4/101/76/748 |
3.9 |
264 |
15/27/ 120 |
1500 |
0.2/0.6 |
SI |
VSR+, TBC |
NO |
Neumonía con derrame pleural |
|
4 |
8.7/3400/ 84 |
6,8/256/98/4707 |
2.3 |
723 |
19/39/ 71 |
1429 |
1.4/2.2 |
SI |
CMV IgM+, , sepsis por Pseudomona |
NO |
Shock séptico |
|
5 |
9.8/6525/ 77 |
4.8/114/190/245 |
3.8 |
142 |
14/35/ 197 |
350 |
0,41/ 0,5 |
SI |
Hepatitis |
NO |
Shock séptico |
|
6 |
9/540/11 |
5,9/40/ 22/681 |
4 |
118 |
16/42/ 182 |
>1500 |
0,38/ 0,4 |
SI |
ITU sec P. aeruginosa, Colonización catéter S. epidermidis |
NO |
Sarcoma histiocítico en paciente LLA B |
|
7 |
6,9/1440/ 68 |
0,2/109/68/1229
|
2 |
194 |
11/25/ 268 |
145 |
0,41/ 0,6 |
NO |
Rinovirus + ITU sec Klebsiella |
NO |
Convulsiones PIMS |
|
8 |
8.5/2640/ 179 |
0,3/43/ 13/516 |
2,4 |
514 |
30,6/32/ 273 |
1044 |
0,18/ 0,3 |
SI |
Parotiditis COVID + Rinovirus + |
NO |
NO |
|
9 |
8.5/920/33 |
12,9/ 144/84 696 |
2,4 |
686 |
14,8/ 41.5/91 |
>1500 |
0,53/ 0,6 |
SI |
- |
SI 47XY |
Hepatitis |
|
10 |
6.9/1600/ 25 |
0,2/59/ 46/622 |
3,7 |
266 |
16/30,4/ 138 |
621 |
0,46/ 0,3 |
NO |
COVID +, VSR, Rinovirus |
NO |
Actividad NK ausente |
|
11 |
8.1/9700/ 42 |
4/124/ 168/619 |
3.5 |
285 |
22/38/ 110 |
610 |
1,1/1,5 |
SI |
Aspergilosis, candidiasis y mucor a foco abdominal |
NO |
Shock séptico enteral Expresión de perforinas disminuida |
|
12 |
7.9/390/ 401 |
2.8/44/ 62/648 |
2,5 |
299 |
67/30/ 105 |
740 |
0,2/0,9 |
NO |
Aspergilosis invasiva |
NO |
FMO |
Tabla 4. Valoración inmunológica
|
Nº |
Expresión de perforinas (CF) |
Expresión SAP (CF) |
Expresión de XIAP (CF) |
Expresión CD107a (CF) |
Biología molecular |
|
5 |
NR |
… |
… |
NR |
Secuenciación gen SH2D1A (SAP) : sin variantes |
|
6 |
Normal |
NR |
NR |
NR |
Panel de desregulación inmune: sin variantes |
|
7 |
NR |
NR |
NR |
NR |
Panel de genes de enfermedades autoinflamatorias y secuenciación de genes de FHLH: sin variantes |
|
8 |
Normal |
Normal |
Normal |
…. |
Panel de genes de inmunodeficiencias primarias: sin variantes |
|
9 |
Normal |
NR |
NR |
Normal |
NR |
|
10 |
NR |
NR |
NR |
NR |
Panel de genes de inmunodeficiencias: en proceso |
|
11 |
Disminuida |
NR |
NR |
NR |
Secuenciación PRF y XIAP: sin variantes |
Gráfico 1. Descripción general del protocolo de tratamiento de HLH-2004

Gráfico 2. Curva de sobrevida global
Conclusiones
La HLH o SHF se caracteriza por la activación patológica del sistema inmune, se presenta tanto en casos con historia familiar de enfermedad o secundario a otros desencadenantes como infecciones, enfermedades hemato-oncológicas y reumatológicas.
No existe ninguna medida preventiva conocida hasta el momento. Es de gran importancia la sospecha clínica precoz, ya que de ello depende el inicio temprano del tratamiento y el pronóstico del paciente.
Se define gracias a una serie de criterios diagnósticos establecidos por la Sociedad de Histiocitosis, entre los que se destacan por su frecuencia las citopenias, la fiebre y la hepatoesplenomegalia. Los estudios disponibles como tamizaje de HLH primario son: expresión de perforinas y CD107a por citometría de flujo, siendo confirmados luego por estudios de biología molecular, ya sea secuenciación génica por Sanger o secuenciación de próxima generación (paneles de genes, secuenciación exómica). Es primordial el comienzo precoz del tratamiento. Sin terapia la mortalidad es muy elevada. En pacientes con formas primarias y alteración genética subyacente conocida, está indicado el consejo genético y la única opción curativa de estos casos es el trasplante alogénico de células progenitoras hematopoyéticas.
Bibliografía
1. Scott R, Robb A. Histiocytic medullary reticulosis. Lancet. 1939; 234(6047):194-8.
2. Farquhar JW, Claireaux AE. Familial hemophagocytic reticulosis. Arch Dis Child. 1952; 27:519-525.
3. Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr. 1985;106(4):561-6.
4. Itziar Astigarraga et al. Síndromes hemofagocíticos: la importancia del diagnóstico y tratamiento precoces. An Pediatric (Barc). 2018;89(2):124 e1-124 e8.
5. Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. 2020;135:1332-43.
6. Sen ES, Steward CG, Ramanan AV. Diagnosing hemophagocytic syndrome. Arch Dis Child. 2017;102:279-84.
7. Janka GE. Linfohistiocitosis hemofagocítica familiar. Eur J Pediatr. 1983; 140:221-2306.
8. Janka GE, Schenider EM. Manejo moderno de niños con linfohistiocitosis hemofagocítica. Br J Haematol. 2004,124:4-14
9. Arceci RJ. When T cells and macrophages do not talk: the hemophagocytic syndromes. Curr Opin Hematol. 2008;15:359-67.
10. Soy M, Atagündüz P, Atagündüz I, Sucak T. Hemophagocytic lymphohistiocytosis: a review inspired by the COVID-19 pandemic. Rheumatol Int. 2021; 41:7-18.
11. Histiocyte Society HLH-2004, Hemophagocytic Lymphohistiocytosis Study Group, Treatment Protocol of the Second International HLH Study 2004, Start of the Study: January 2004 Chairman: Jan-Inge Henter, MD, PhD, Stockholm, Sweden.
12. Imashuko S. Clinical features and treatment strategies of Epstein-Barr virus associated hemophagocytic lymphohistiocytosis. Crit Rev Oncol Hematol. 2002;44:259-72.
13. Sen ES, Steward CG, Ramanan AV. Diagnosing haemophagocytic syndrome. Arch Dis Child. 2017;102:279-84.