Descripción y evolución del primer caso de síndrome de Jacobsen diagnosticado en Argentina, su analogía con anemia de Fanconi
Description and evolution of the first case of Jacobsen's syndrome in Argentina, its analogy with Fanconi anemia
Benasayag S1, Goncalves AR1, Laiseca J1, Serale C1, Lopez S1, Galesi O2, Mattina T3, Mohamad P1
1Fundagen, Centro de Genética Integral, CABA, Argentina
2Laboratory of Medical Genetics, Oasi Research Institute-IRCCS, Italia
3Centro Regional de Referencia de Catania, Italia
ORCID ID
Benasayag S: 0009-0006-9054-8053
Goncalves AR: 0009-0009-4099-0516
Laiseca J: 0009-0004-5163-5572
Serale C: 0009-0007-6366-6466
Lopez S: 0009-0006-0926-3941
Galesi O: 0000-0002-7877-938X
Mattina T: 0000-0001-6143-6682
Mohamad P: 0009-0004-2018-7885
Palabras clave: pancitopenia,
anemia de Fanconi,
síndrome de Jacobsen.
Keywords: pancytopenia,
Fanconi anemia,
Jacobsen syndrome.
Resumen
Descripción del primer caso de síndrome de Jacobsen en Argentina y su evolución. Se evaluó una paciente de un año y medio con pancitopenia congénita, dismorfias y manchas en la piel. Se descartaron causas infecciosas y toxicológicas. Inicialmente se sospechó de anemia de Fanconi (AF). El test de diepoxibutano (DEB) dio negativo, ecocardiograma y radiografías normales. En el cariotipo se observó una deleción de novo en la región 11q23 compatible con el síndrome de Jacobsen. Este síndrome es causado por una deleción de genes contiguos y se caracteriza por déficit intelectual, retraso del crecimiento pre y postnatal, rasgos faciales característicos, trombocitopenia o pancitopenia. Además pueden presentar malformaciones del corazón, riñón, tracto gastrointestinal, genitales, sistema nervioso central, esqueléticas y anomalías inmunológicas. Se realizó el estudio de micromatriz precisando el punto de ruptura en la región 11q24.1 y el tamaño de la deleción de 13,133 Mb.
Abstract
Description of the first case of Jacobsen syndrome in Argentina and its evolution. A one and a half year old patient with congenital pancytopenia, dysmorphia and skin spots was evaluated. Infectious and toxicological causes were ruled out. Fanconi anemia was initially suspected. The diepoxybutane test (DEB) was negative, normal echocardiogram and X-rays. The karyotype revealed a de novo deletion in the 11q23 region, compatible with Jacobsen's syndrome. This syndrome is caused by a deletion of contiguous genes and is characterized by intellectual deficit, prenatal and postnatal growth retardation, characteristic facial features, thrombocytopenia or pancytopenia. In addition, they can present malformations of the heart, kidney, gastrointestinal tract, genitalia, central nervous system, skeletal and immunological abnormalities. The microarray study was performed specifying the breakpoint in the 11q24.1 region and the size of the deletion of 13,133 Mb.
Introducción
La pancitopenia (disminución del valor por debajo de lo normal para la edad de todas las series sanguíneas) es un frecuente motivo de consulta en Hematología y Genética. Las causas incluyen: trastornos autoinmunes, fallos medulares, patologías genéticas, infecciosas, desnutrición, déficit de vitamina B12 y ácido fólico, hiperesplenismo, embarazo, tratamientos del cáncer o exposición a ciertas toxinas, sustancias químicas o medicamentos.
La anemia de Fanconi (AF) es un trastorno multisistémico de base genética y poco frecuente caracterizado por pancitopenia progresiva con insuficiencia medular, malformaciones congénitas variables y predisposición al desarrollo de tumores hematológicos o sólidos. La AF es genéticamente heterogénea y las variantes implicadas están asociadas a la reparación del ADN y a la estabilidad genómica. Más del 90% de los pacientes presenta mutaciones de los genes FANCA, FANCC, FANCG o FANCD2. El 10% restante se reparte entre otros 18 genes descritos(1-3).
El síndrome de Jacobsen es un trastorno de genes contiguos que da lugar a anomalías congénitas y discapacidad intelectual causado por una pérdida o deleción parcial en el brazo largo del cromosoma 11. Las características clínicas más comunes incluyen retraso del crecimiento pre y postnatal, retraso del desarrollo psicomotor, rasgos faciales característicos, malformaciones del corazón, riñón, tracto gastrointestinal, genitales, sistema nervioso central, esqueléticas(4) y anomalías inmunológicas(5). Alrededor del 50 % de los pacientes con deleción 11q y síndrome de Jacobsen también muestran problemas de conducta compatibles con el trastorno del espectro autista(6).
El objetivo de este trabajo es describir cómo se arribó al diagnóstico y la evolución clínica de una paciente con pancitopenia y sospecha de anemia de Fanconi que reveló una anomalía cromosómica definiendo el primer caso de síndrome de Jacobsen en Argentina.
Materiales y métodos
Paciente femenina de 14 meses que concurrió a la consulta con Genética por derivación de la hematóloga. Nació a las 32 semanas de embarazo, con un peso de 1200 gr. En Neonatología presentó un cuadro compatible con sepsis por trombocitopenia severa y sangrado intestinal. Requirió transfusión de plaquetas y tres transfusiones de glóbulos rojos por anemia severa sintomática. A los 3 meses (al alta de Neonatología) consultó a Hematología presentando pancitopenia con leucopenia y plaquetopenia moderadas, anemia arregenerativa y macrocítica, sin requerimiento transfusional y en tratamiento con vitamina B12 y ácido fólico por vía oral. Con los meses evolucionó hacia la estabilidad de su plaquetopenia.
Se observó mejoría del recuento reticulocitario, plaquetario, neutropenia, retraso madurativo moderado, mal progreso de peso y facies peculiar por lo que fue derivada a Genética.
Al examen físico se observó frente prominente, orejas de implantación baja con rotación posterior, hipertelorismo, labios finos, epicanto, mácula rosácea en región frontal, cejas despobladas, nariz larga, filtrum liso, cara triangular y mácula café con leche de 1 x 1 cm en antebrazo derecho. Inicialmente impresionaba posición anormal del pulgar, lo cual junto con la mácula y la pancitopenia generaron la sospecha diagnóstica de anemia de Fanconi (Figura 1).
Estudios solicitados
Laboratorio. Debido a la sepsis se estudiaron posibles infecciones virales y se evaluó también genotoxicidad. Se realizó un laboratorio de rutina, serología para toxoplasmosis, Chagas, sífilis, hepatitis B, C y A, VIH, Epstein Barr, citomegalovirus y parvovirus.
Para evaluar el posible diagnóstico de anemia de Fanconi se realizaron: radiografía de mano, ecografía abdominal y renal, doppler de vasos abdominales y ecocardiograma. Se solicitaron en Genética los siguientes estudios: el test de DEB para evaluar anemia de Fanconi(7), test de micronúcleos para evaluación de genotoxicidad(8) y estudio citogenético con la técnica de Moorhead modificado(9). Este último se realizó por duplicado en dos cultivos de linfocitos en sangre periférica estimulados con fitohemaglutinina y posterior técnica de bandeo GTG. También se evaluaron cariotipos parentales. Posteriormente se solicitó el estudio de hibridización in-situ fluorescente (FISH) del gen MLL utilizando la sonda break apart de Lexel Médica(10).
A fin del año 2020 se logró obtener el estudio de micromatriz de la paciente. El mismo fue realizado en la unidad de Diagnóstico y Tratamiento de Enfermedades Genéticas del Centro Regional de Referencia de Catania, Italia.
El estudio de micromatriz se realizó en ADN genómico aislado de linfocitos de sangre periférica que se amplificó, fragmentó e hibridizó de acuerdo con las instrucciones del fabricante utilizando el estándar Agilent SurePrint G3 Human CGH+SNP 4 × 180K Microarray (Agilent Technologies, California, EE. UU.), con una mediana general de espacio entre sondas de 25,3 Kb. La imagen de la matriz se adquirió con el escáner láser de Agilent G5761A (Agilent Technologies, California, EE. UU.) y fue analizado con el programa de Agilent Cytogenomics (v.5.0.2.5). Las coordenadas genómicas se informaron de acuerdo con el genoma de referencia GRCh37/hg19.
Resultados
Todos los resultados serológicos dieron negativos (toxoplasmosis, VDRL, HBV, HCV, HAV, HIV, EBV, CMV y parvovirus). Las imágenes también fueron normales (radiografía de mano, ecografía abdominal, renal, vesical y de vasos abdominales, así como el ecocardiograma).
El test de DEB dio un valor normal (0,18 vs 0,07 rup/cél) descartando el diagnóstico de anemia de Fanconi.
El test de micronúcleos arrojó un resultado negativo descartando genotoxicidad.
El estudio citogenético reveló un cariotipo patológico con una deleción en el brazo largo del cromosoma 11 a nivel de la región q23 en un total de 20 células analizadas (Figura 2). Para confirmar que la deleción hallada no fuera heredada se realizaron cariotipos parentales los cuales resultaron normales (cariotipo de la madre: 46,XX[20]; cariotipo del padre: 46,XY[20]).
Debido a que en la región involucrada se encuentra el gen MLL, responsable del 6% de los casos de leucemia linfocítica aguda (LLA) se quiso descartar alteración en el mismo. El resultado del estudio de FISH para el gen MLL dio normal en 400 núcleos interfásicos (nuc.ish(MLLx2)[400]).
El estudio de micromatriz precisó el punto de ruptura en la región 11q24.1 y el tamaño de la deleción de 13,133 Mb (ArrGRCh37/[hg19] 11q24.1-q25(121.801.090-134.934.196)x1 tamaño: 13.133.106 pb) (Figura 3).
Se compararon los estudios y el fenotipo de la paciente contra un paciente con anemia de Fanconi. La AF se caracteriza por anomalías físicas, falla de la médula ósea y mayor riesgo de cáncer. Las anomalías físicas, presentes en aproximadamente el 75% de las personas afectadas, incluyen una o más de las siguientes anormalidades: estatura baja, pigmentación anormal de la piel, malformaciones esqueléticas de las extremidades, microcefalia y anomalías oftalmológicas y genitourinarias. La insuficiencia progresiva de la médula ósea con pancitopenia se presenta típicamente en la primera década, a menudo inicialmente con trombocitopenia o leucopenia. La incidencia de leucemia mieloide aguda es del 13% a los 50 años. Los tumores sólidos, particularmente de cabeza y cuello, piel y tracto genitourinario son más comunes en personas con anemia de Fanconi (Tabla 1).
Dada la alteración citogenética encontrada en la región 11q23 sumada al fenotipo peculiar se reveló que estábamos frente al primer caso de síndrome de Jacobsen de la Argentina (2013).
Luego de 5 años del diagnóstico la niña evolucionó con múltiples infecciones de causa respiratoria, rinorrea crónica, broncoespasmos a repetición y dermatitis atópica, por lo cual se evaluó por Inmunología. Desde el aspecto hematológico se diagnosticó hipoplasia medular por biopsia de médula ósea con pancitopenia moderada. Se detectó hipogammaglobulinemia (IgA 38, IgG 487 e IgM 18), linfopenia global con inversión CD4/CD8 (CD4 143, CD8 205) y linfopenia CD4 severa. Se diagnosticó como inmunodeficiencia en tratamiento con profilaxis antibiótica con buenos resultados. Actualmente se encuentra en seguimiento por los Servicios de Inmunología, Genética, Pediatría y Hematología, recibe tratamiento interdisciplinario en el área psicológica desde el año 2014 y realiza distintas terapias de estimulación artística y cognitiva como equinoterapia, musicoterapia y natación. (Figura 4).
Actualmente la niña aprendió a colaborar con el proceso de vestimenta, controla esfínteres, se conecta con el entorno, comunica sus necesidades, tiene producción verbal, presta atención cuando se le habla, sostiene la mirada, acepta límites, ofrece disculpas y puede manejarse en la vía pública.
Discusión
El síndrome de Jacobsen es un trastorno de genes contiguos que da lugar a anomalías congénitas y discapacidad intelectual causado por una pérdida o deleción parcial en el brazo largo del cromosoma 11. Hasta el momento, se han descrito alrededor de 300 casos. Se estima una prevalencia de 1/100.000 nacimientos, con una tasa mujer/hombre de 2:1(11,12).
Normalmente se presenta al nacimiento como una trombocitopenia o pancitopenia. Pueden presentar malformaciones del corazón, riñón, tracto gastrointestinal, genitales, sistema nervioso central y esqueléticas. También anomalías inmunológicas, hormonales, de la audición y oculares. Las anomalías cardíacas pueden ser muy graves y requerir intervención quirúrgica en el período neonatal. Alrededor de un 20% de los niños fallecen durante los dos primeros años de vida, principalmente por complicaciones de la cardiopatía congénita y, con menor frecuencia, por complicaciones hemorrágicas(12).
El tamaño de la deleción varía de 7 a 20 Mb, con el punto de ruptura proximal en la sub-banda 11q23.3, extendiéndose generalmente hasta el final del cromosoma. Esta deleción es de novo en el 85 % de los casos y en el 15% restante puede deberse a una segregación desbalanceada de una translocación familiar recíproca balanceada o a otros reordenamientos cromosómicos.
Se observa una gran diversidad de fenotipos en los pacientes reportados(13). Si bien inicialmente se creía que las deleciones más grandes generaban cuadros clínicos más severos, la deleción en nuestra paciente (que es de 13,3 Mb e incluye unos 140 genes descritos hasta el momento) es bastante extensa para el cuadro clínico que presenta. Si bien tiene autismo, dice algunas palabras, camina, puede relacionarse, interactuar y realizar tareas de la vida cotidiana.
Los resultados resaltan la importancia de realizar estudios de citogenética clásica para descartar anemia de Fanconi, en todos aquellos niños que padecen pancitopenia, rasgos dismórficos y/o discapacidad intelectual. Esto permitiría detectar anomalías estructurales que llevan a la identificación de posibles síndromes genéticos, permitiendo un adecuado asesoramiento personal y familiar.
Luego de 9 años la paciente se encuentra en buen estado general, realizando múltiples terapias de estimulación (fonoaudiología, musicoterapia, equinoterapia y natación) las cuales junto con el apoyo, contención y aceptación familiar permiten tanto una evolución clínica favorable como una mejor calidad de vida.
En junio de 2018 se crea la Fundación 11q Latinoamérica-Síndrome de Jacobsen.
Hasta el momento de esta publicación se han descrito en Latinoamérica: 10 casos en Argentina, 10 en Colombia, 4 en Chile, 4 en Uruguay, 6 en Perú, 5 en México , 2 en Ecuador, 2 en Venezuela, 3 en Guatemala, 1 en Cuba y 35 en Brasil.
Este estudio resalta la importancia del trabajo de un equipo multidisciplinario así como la interacción conjunta de la familia y el equipo de salud para un mejor asesoramiento y el bienestar del paciente.
Agradecimientos.
A Cecilia Schiaretti por los aportes y la colaboración constante para la realización de este trabajo y a Yamila Trinchero y Gisela Suárez por el trabajo técnico realizado.
Bibliografía
1. Sagaseta de Ilurdoz M, Molina J, Lezáun I et al. Updating Fanconi's anaemia. An Sist Sanit Navar. 2003 Jan-Apr;26(1):63-78.
2. Green AM, Kupfer GM. Fanconi anemia. Hematol Oncol Clin North Am. 2009 Apr;23(2):193-214.
3. Mehta PA, Ebens C. Fanconi Anemia. 2002 Feb 14 [Updated 2021 Jun 3]. In:Adam MP, Mirzaa GM, Pagon RA et al., ed. GeneReviews® [Internet].Seattle (WA): University of Washington, Seattle; 1993-2022.
4. Fernández González N, Prieto Espuñes S, Ibáñez Fernández A et al. Deleción terminal del 11q (síndrome de Jacobsen) asociada a atresia duodenal con páncreas anular. Anales de Pediatría. 2006;65(3):249-252.
5. Dalm VA, Driessen GJ, Barendregt BH, van Hagen PM, van der Burg M. The 11q Terminal Deletion Disorder Jacobsen Syndrome is a Syndromic Primary Immunodeficiency. J Clin Immunol. 2015 Nov;35(8):761-8.
6. Natacha Akshoomoff, Sarah N. Mattson, Paul D. Grossfeld. Evidence for autism spectrum disorder in Jacobsen syndrome: identification of a candidate gene in distal 11q. Genet Med. 2015 Feb;17(2):143-8.
7. Auerbach AD. Diagnosis of Fanconi Anemia by Diepoxybutane Analysis. Curr Protoc Hum Genet, 2015. Apr 1;85:8.7.1-17.
8. Zalacain M, Sierrasesúmaga L, Patiño A. El ensayo de micronúcleos como medida de inestabilidad genética inducida por agentes genotóxicos. Sis San Navarra. 2005;28(2).
9. Benasayag S, Gallino I. Bases citogenéticas para la práctica hematológica. De lo supuesto a lo expuesto en nomenclatura citogenética. Hematología. 2010;14(2):58-68.
10. Mc Gowan-Jordan J, Hasting RJ, Moore S. Fish MLL. ISCN 2020. An international System for Human Cytogenetic Nomenclature (2020). Karger ed, cap. 9, p53.
11. Grossfeld PD, Mattina T, Lai Z et al. The 11q terminal deletion disorder: a prospective study of 110 cases. Am J Med Genet A. 2004 Aug 15;129A(1):51-61.
12. Mattina T, Perrotta CS, Grossfeld P. Jacobsen Syndrome. Orphanet J Rare Dis. 2009 Mar 7;4:9.
13. Favier R, Akshoomoff N, Mattson S, Grossfeld P. Jacobsen syndrome: Advances in our knowledge of phenotype and genotype. Am J Med Genet C Semin Med Genet. 2015 Sep;169(3):239-50.
Tabla 1. Cuadro comparativo entre patologías según la paciente en estudio.
|
|
Síndrome de Jacobsen |
Anemia Fanconi |
|
Incidencia |
1/100000 |
1/160000 |
|
Etiología |
Deleción genes contiguos |
Multigénica |
|
Herencia |
Autosómica dominante/de novo |
recesiva |
|
Facies peculiares |
SÍ |
SÍ |
|
Trigonocefalia y dismorfia facial |
SÍ |
NO (otras) |
|
Retraso psicomotor |
SÍ |
SÍ (16%) |
|
Alteraciones esqueléticas |
NO |
SÍ |
|
Pigmentación anormal |
NO/SÍ |
SÍ |
|
Defectos cardíacos |
SÍ |
SÍ (13%) |
|
Alteraciones hematológicas |
SÍ |
SÍ |
|
Alteraciones cromosómicas |
del(11)(q23) |
NO |
|
Sitio frágil 11B |
SÍ |
NO |
|
Rupturas / Testde DEB |
NEGATIVO |
POSITIVO |
|
Genes de reparación |
NO |
(13 genes FANC) |
|
Desarrollo a LMA |
NO |
SÍ |
Síndrome de Jacobsen Anemia de Fanconi

Figura 1. Fotos de la paciente en estudio (Jacobsen) a la izquierda vs anemia de Fanconi. Facies de paciente con Jacobsen: frente prominente, hipertelorismo, labios finos, epicanto, mácula rosácea en región frontal, cejas despobladas, nariz larga, filtrum liso, cara triangular. B. Facies de paciente con anemia de Fanconi: frente prominente, hipotelorismo, nariz de base ancha, cejas despobladas, labios finos, filtrum liso y corto, orejas de implantación baja. C. Mácula hipercrómica en paciente con Jacobsen. D. Mácula hipercrómica en paciente con anemia de Fanconi la cual es más grande e hiperpigmentada. E. mano de paciente con Jacobsen, se observaba leve desviación del pulgar la cual resultó ser posicional y no se condecía con alteración en las radiografías. F. Mano de paciente con anemia de Fanconi en la cual se observa clara desviación cubital del pulgar.
 Figura
2. Cariotipo de la paciente en estudio 46,XX,del(11)(q23)[20]. Se indica la
deleción del cromosoma 11 con flecha.
Figura
2. Cariotipo de la paciente en estudio 46,XX,del(11)(q23)[20]. Se indica la
deleción del cromosoma 11 con flecha.
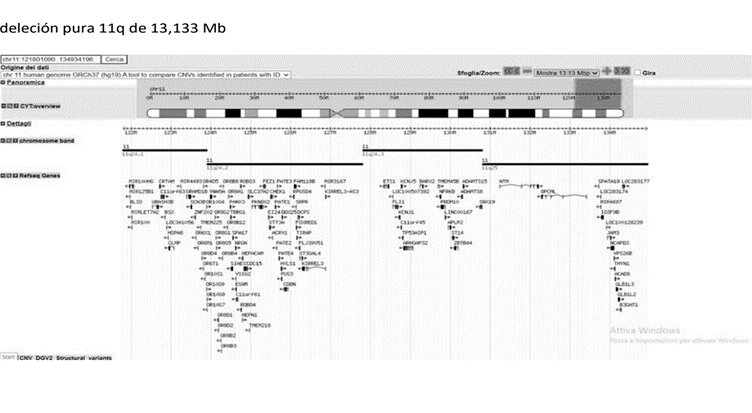
Figura 3. Segmento representativo de la deleción del cromosoma 11 a nivel de la región q24.1 con un tamaño de 13,133 Mb. Se incluyen en la figura algunos de los genes delecionados.

Figura 4. Foto de la paciente a los 8 años de edad.