Histiocitosis de células de Langerhans en adultos. Reporte de 2 casos
Langerhans cell histiocytosis in adults. Report of 2 cases
Escalera S1; Alfonso G1; Freitas J1; Maymo D1; Vigna C2
1 Servicio Hematología - Hospital A. Posadas - Buenos Aires, Argentina
2 Servicio Anatomía Patológica - Hospital A. Posadas - Buenos Aires, Argentina
Palabras clave: histiocitosis,
células de Langerhans,
mieloide.
Keywords: histiocytosis,
Langerhans cell,
myeloid.
Resumen
La histiocitosis de células de Langerhans (HCL) es una neoplasia mieloide de células dendríticas (CD) caracterizada por la expansión clonal de precursores mieloides que expresan los antígenos de superficie CD1a+ y CD207+ (langerina). La HCL es una enfermedad rara e infrecuente, de aparición predominantemente pediátrica con una incidencia anual que oscila entre 2 y 9 casos por millón, siendo aun más rara en adultos, con una incidencia anual estimada de aproximadamente 1 caso por millón, aunque probablemente este valor reducido sea a consecuencia de una enfermedad subdiagnosticada en esta población.
El curso clínico es heterogéneo, puede presentarse desde una lesión indolente autolimitada (granuloma eosinofílico solitario) hasta una enfermedad diseminada con disfunción orgánica rápidamente progresiva, que puede llevar a la muerte. La Histiocyte Society (HS) clasifica las formas clínicas de HCL según el número y el tipo de órganos afectados: HCL de un solo sistema (HCL-SS) si está afectado un órgano/sistema (ya sea unifocal o multifocal) y HCL multisistémica (HCL-MS) si hay dos o más órganos/sistemas que estén involucrados. Presentaremos dos casos de HCL uno con compromiso de un SS y otro MS.
Abstract
Langerhans cell histiocytosis (LCH) is a myeloid neoplasm of dendritic cells (DC), characterized by the clonal expansion of myeloid precursors that express the surface antigens CD1a+ and CD207+. LCH is a rare and infrequent disease, of predominantly pediatric onset with an annual incidence ranging between 2 and 9 cases per million, being even rarer in adults, with an estimated annual incidence of approximately 1 case per million, although this value probably reduced is due to an underdiagnosed disease in this population.
The clinical course is heterogeneous, it can present from an indolent self-limited lesion (solitary eosinophilic granuloma) to a disseminated disease with rapidly progressive organ dysfunction, which can lead to death. The Histiocyte Society (HS) classifies the clinical forms of LCH according to the number and type of organs affected: single system LCH (SS-LCH) if one organ/system is affected (either unifocal or multifocal) and multisystem LCH (MS- LCH) if two or more organ systems are involved. We will present two cases of HCL, one with involvement of a SS and another MS.
Introducción
La histiocitosis de células de Langerhans (HCL) es una neoplasia mieloide de células dendríticas (CD), caracterizada por la expansión clonal de precursores mieloides que expresan los antígenos de superficie CD1a+ y CD207+(1-3). La HCL fue uno de los primeros trastornos histiocíticos reconocidos como neoplasia hematopoyética por la Organización Mundial de la Salud (OMS) debido al establecimiento de la clonalidad(3).
La HCL es una enfermedad rara e infrecuente, de aparición predominantemente pediátrica, es en esta población de donde derivan la mayoría de los estudios clínicos, con una escasez de datos que examinen a sus contrapartes adultas. A continuación presentaremos dos casos de HCL uno con compromiso de un SS y otro MS(3).
Presentación de casos clínicos
Caso 1
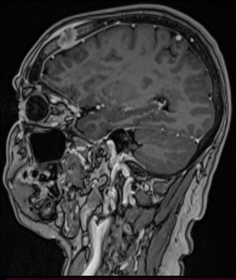
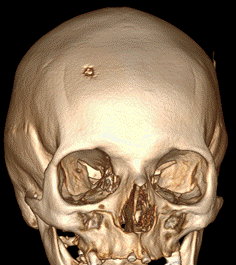
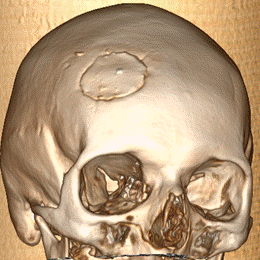
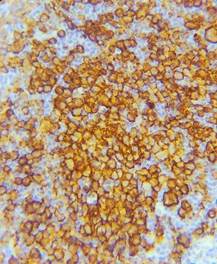
Femenina de 51 años sin antecedentes de relevancia consulta por dolor y tumefacción en región de cuero cabelludo frontal-temporal derecho. Niega traumatismo. Al examen físico presenta en región frontotemporal derecha, lesión indurada dolorosa. Se realiza laboratorio sin particularidades, resonancia magnética nuclear (RMN) de cabeza que evidencia a nivel de la calota frontal derecha imagen nodular mal definida con compromiso de la tabla interna y externa sin compromiso aparente del parénquima cerebral (Figura 1) que es interpretada en primera instancia como una metástasis con primario desconocido. Se solicita tomografía computada (TC) de cuello/tórax/abdomen/pelvis sin evidencia de lesiones sugerentes de neoplasia, por lo que se decide biopsiar dicha lesión. Se realiza exéresis completa de la lesión siendo el resultado de anatomía patológica compatible con HCL (Figura 2. CD1a: positivo; S100: positivo; langerina: positivo). Se realiza PET/TC sin evidencia de captación patológica de FDG, por lo que se interpreta como HCL SS con lesión ósea única (monostótica). Evoluciona favorablemente con episodios de cefalea intermitente que ceden con AINES. Actualmente 10 meses después se encuentra en buen estado general, asintomática. Se realiza RMN de control sin progresión de lesión ni aparición de lesiones nuevas.

![]()
![]()
![]()
![]()
Figura 1. RMN de cabeza. A. En calota frontal derecha imagen nodular mal definida con compromiso de la tabla interna y externa regional, impresiona insinuarse sobre el espacio extradural adyacente sin compromiso aparente del parénquima cerebral. B. Reconstrucción ósea C y D. Post operatorio a 6 meses


![]()
![]()
![]()
Figura 2. A. CD1a: positivo B. S100: positivo C. Langerina: CD207+
Caso 2
Femenina de 30 años sin antecedentes de relevancia. Consulta repetidamente desde el 2016 por deterioro progresivo de la agudeza visual agregando posteriormente polidipsia, poliuria y cambios conductuales. En 2017 se diagnostica diabetes insípida por lo que se realiza RMN de hipófisis que evidencia una imagen a nivel del quiasma óptico (Figura 3), con hallazgos compatibles con proceso infiltrativo. Por motivos personales y de cobertura médica no se realiza biopsia de dicha lesión, quedando con conducta expectante. En 2019 presenta cuadrantanopsia heterónima bitemporal inferior y aparecen lesiones ulceradas en genitales y periné, por las cuales presenta repetidas internaciones donde recibe múltiples esquemas antimicrobianos sin mejoría por lo que se realiza biopsia de dichas lesiones que informa proliferación difusa de linfocitos y abundante cantidad de histiocitos. CD 68: positivo en histiocitos; vimentina: positivo; CD3 (pan T): positivo en linfocitos de fondo; S-100: positivo; el perfil inmunológico corresponden a una HCL. Realiza tratamiento con corticoides tópicos y orales con mejoría parcial (Figura 5). Desde el 2020 pierde seguimiento por la pandemia de COVID 19 recibiendo solo terapia hormonal sustitutiva (DBT insípida e hipotiroidismo). En junio del 2022 acude a endocrinología por presentar durante el último mes amaurosis bilateral asociada a fiebre intermitente, dermatitis seborreica y deterioro del estado general. Por antecedente de HCL cutánea la derivan al Servicio de Hematología. Al momento de la consulta familiar refiere que el cuadro se exacerba con episodios de sensorio alternante y discurso incoherente, por lo que se realiza RMN de encéfalo que evidencia crecimiento de lesión hipotalámica asociada a marcado edemas y múltiples lesiones que comprometen bulbo y protuberancia (Figura 4). Debido al gran compromiso del sistema nervioso central (SNC) (Figura 4) asociado a hallazgos pulmonares (Figura 7) e histología cutánea con HCL se realiza ateneo multidisciplinario donde se interpreta cuadro como HCL con compromiso multisistémico de alto riesgo y se decide inicio de QMT con ciclos de arabinósido de citosina (ARA-C) + dexametasona. Realiza 3 ciclos de AraC + dexametasona con mejoría clínica e imagenológica (Figuras 5 y 6B).
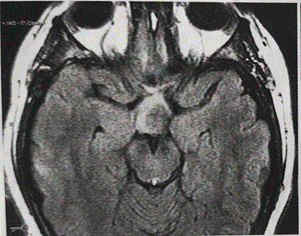

Figura 3. RMN de hipófisis 01/2017. Imagen hipointensa en T1 e isointensa en T2 que realza con contraste de forma heterogénea a nivel del quiasma óptico, de aproximadamente 22 mm x 11 mm x 15 mm, con tallo pituitario y glándula hipófisis respetados.


Figura 4. RMN de encéfalo 06/2022. En región supraselar imagen de aproximadamente 38 mm x 28 mm x 41 mm rodeada de edema que genera efecto de masa sobre el piso del tercer ventrículo. Múltiples imágenes nodulares con refuerzo homogéneo post contraste y edema perilesional a nivel del bulbo y protuberancia.


Figura 5. RMN de encéfalo 08/22. Control posterior a 3 ciclos de QMT. Se observa persistencia de imagen supraselar en menor tamaño de aproximadamente 32 mm x 16 mm x 30 mm con disminución del edema.


![]()
![]()
Figura 6. A. Úlcera en nido en ingle izquierda. B. Misma lesión después de 3 ciclos de QMT.
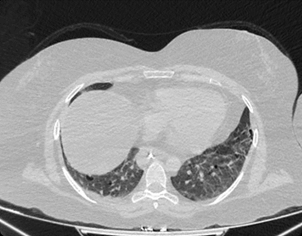
Figura 7. TC de tórax 06/2022. A. Se observan a nivel de ambas bases pulmonares aisladas imágenes con patrón quístico parenquimatoso de distribución centroacinar.
Discusión
La histiocitosis de células de Langerhans (HCL) es una neoplasia mieloide de células dendríticas (CD) patológicas, caracterizada por la expansión clonal de precursores mieloides que expresan los antígenos de superficie CD1a+ y CD207+(1-3). La clasificación de neoplasia mieloide se debe a la identificación de mutaciones recurrentes en BRAF-V600E que representan alrededor del 50% de los casos. Las mutaciones en la proteína cinasa 1 activada por mitógenos (MAP2K1) y los genes del homólogo A del oncogén viral del sarcoma murino v-Raf (ARAF) también se han asociado recientemente con la HCL(3,4). La acumulación de estas células patológicas mezcladas con linfocitos, macrófagos, eosinófilos y neutrófilos provoca un infiltrado inflamatorio que se evidencia en las clásicas lesiones, que pueden surgir en prácticamente cualquier sistema de órganos (sin inducir necesariamente disfunción), pero tienen una afinidad particular por los huesos, la piel, los pulmones y la hipófisis(2,4,5).
La HCL en adultos cursa predominantemente con compromiso multisistémico (68,6%), mientras que la enfermedad monosistémica representa el 31,4%(6). Las lesiones óseas más frecuentes se encuentran en el cráneo (55%), columna vertebral (30%), mandíbula (26%), pelvis (20%), costillas (18%), piernas (18%), mano (13%) y hombro (12%). Estas lesiones se manifiestan con dolor focal y crecimiento anormal del tejido blando adyacente al hueso afectado y al examen clínico suele revelar una protuberancia blanda y sensible(5).
La HCL pulmonar (PLCH, por sus siglas en inglés) puede ocurrir como parte de una HCL multisistémica o, más comúnmente, como una enfermedad aislada (PLCH de un solo sistema). Se ha informado que ocurre hasta en el 35% de los pacientes y que la afectación pulmonar aislada en adultos está relacionada con el hábito tabáquico. Los hallazgos radiográficos son típicos de la presencia de un patrón reticulonodular con formación quística(3,4).
Las lesiones cutáneas representan un síntoma muy frecuente de consulta, éstas aparecen como nódulos, placas, úlceras, costras, granulomas o erupciones parecidas a la dermatitis seborreica generalizada. Las regiones más comúnmente afectadas son la mucosa oral, genital así como también áreas de fricción como las ingles y axilas(4).
La HCL del SNC se puede dividir en: (1) lesiones de masa focal y (2) lesiones asociadas con neurodegeneración progresiva. Las regiones comúnmente afectadas por lesiones de masa son hipófisis, paquimeninges, plexo coroideo, glándula pineal y el parénquima cerebral(3,4,7,8). La infiltración de la hipófisis se presenta clínicamente con endocrinopatías donde la más frecuente es la diabetes insípida (DI) llegando a afectar hasta un 29% de pacientes con HCL. La DI es la presentación inicial más común que puede preceder a un diagnóstico de HCL por meses o incluso años(7.8). La HCL neurodegenerativa (LCH-ND) puede presentarse clínicamente como: a) imágenes anormales del SNC asociadas con HCL (LCH-associated abnormal CNS imaging/LACI), donde el paciente presenta imágenes patológicas pero se encuentra asintomático; b) síntomas anormales del SNC asociados con HCL (LCH-associated abnormal CNS symptoms/LACS), que incluye hallazgos radiológicos y síntomas que varían según el área afectada (temblores, alteración de la marcha, espasticidad motora, ataxia, disartria, disfagia, cambios de comportamiento, trastornos del aprendizaje o problemas psiquiátricos)(3,4,8).
El diagnóstico debe incluir un interrogatorio exhaustivo y un examen físico detallado en función de evaluar posibles sitios de infiltración orgánica. El patrón oro para el diagnóstico de HCL es la biopsia de lesión asociada a técnicas de inmunohistoquímica (CD1a+ y CD207+). Se recomienda realizar un laboratorio completo (hemograma, ionograma, hepatograma, PCR, perfil tiroideo), PET/TC de cuerpo entero, RMN de encéfalo, si presenta anomalías en el hemograma biopsia de médula ósea y, si se encuentran disponibles, estudios mutacionales del BRAF-V600E.
El enfoque terapéutico debe ser personalizado y en función del compromiso del órgano afectado. El tratamiento de HCL-SS ósea unifocal depende de la ubicación de la lesión y los síntomas que produce. Se puede realizar exéresis quirúrgica completa, inyección intralesional de corticoides y/o radioterapia. En la HCL-SS ósea multifocal se recomienda el uso de bifosfonatos por su efecto antiosteoclástico que ayuda a reducir las sustancias inflamatorias nocivas y otras citoquinas degradantes en las lesiones óseas activas. La radioterapia local representa una opción terapéutica en los casos de falta de respuesta al uso de bifosfonatos. En caso de lesiones refractarias se recomienda el uso de quimioterapia sistémica con arabinósido de citosina (ARA-C), ya que este se considera como el régimen más eficaz y menos tóxico(2).
En la PLCH el cese del hábito tabáquico ha demostrado casos de remisión completa, por lo que actualmente se recomienda en pacientes con dicha presentación el abandono del tabaco como terapia de primera línea.
El tratamiento para la HCL-MS no se encuentra estandarizado, muchos de los esquemas terapéuticos para adultos se extrapolan de esquemas pediátricos donde se observó que el uso de vinblastina + dexametasona en adultos conllevaba altas tasas de recaída con un perfil de toxicidad más alto. Actualmente el uso de citarabina (AraC 100 mg/m2 días 1-5 subcutánea cada 21 días por 6 meses) o cladribine (2-CdA 5 mg/m2/d por 5 días cada 24 días por 6 meses) son considerados de primera línea dado que demostraron lograr mayores respuestas, menos recaídas y menor toxicidad(3,5).
Con la mejor comprensión de la enfermedad a partir de la identificación de mutaciones moleculares se desarrollaron enfoques terapéuticos novedosos basados en inhibidores de BRAF (vemurafenib, dabrafenib) que han demostrado tasas de respuesta altas en pacientes refractarios a quimioterapia portadores de dicha mutación. Sin embargo, está terapéutica aún no ha sido estandarizada y se necesitan estudios clínicos que avalen su uso en esta población.
En pacientes pediátricos recaídos/refractarios se puede considerar el trasplante de células progenitoras hematopoyéticas (TPH) autólogo o alogénico, pero en adultos la eficacia y las indicaciones siguen sin determinarse.
No están establecidos los criterios de respuesta en adultos, pero el consenso de expertos recomienda estrategias de seguimiento personalizadas cada 3 a 6 meses basadas en el órgano involucrado y tipo de tratamiento utilizado(3).
Conclusiones
Se presentaron dos casos clínicos con dos presentaciones diferentes de la misma enfermedad. El primer caso fue un hallazgo postquirúrgico de la exéresis de una lesión solitaria que actualmente continúa en remisión y no requirió nuevos esquemas terapéuticos. El segundo caso fue el de una mujer joven que, a pesar de realizar múltiples consultas con síntomas inespecíficos pero característicos de la enfermedad y aun presentando hallazgos histopatológicos compatibles con HCL, se retrasó el inicio del tratamiento por más de 5 años repercutiendo en secuelas permanentes que conllevaron a una discapacidad visual definitiva.
En resumen, la HCL sigue siendo una entidad extremadamente rara en adultos. Con frecuencia se presenta como una enfermedad multisistémica con compromiso de órganos de riesgo, por lo cual una demora diagnóstica involucra una terapéutica tardía que deriva en un deterioro de la calidad de vida del paciente.
El pronóstico se correlaciona con la extensión de la enfermedad y el grado de disfunción orgánica, por lo que la elección del tratamiento debe ser individualizada. Aunque los esquemas actuales han demostrado eficacia, se debe continuar en la búsqueda de nuevas dianas terapéuticas que proporcionen mejores resultados y variedad de opciones de tratamiento.
Bibliografía
1. Allen CE, Merad M, McClain KL. Langerhans-Cell Histiocytosis. N Engl J Med. 2018 Aug 30;379(9):856-868.
2. Georgakopoulou D, Anastasilakis AD, Makras P. Adult Langerhans Cell Histiocytosis and the Skeleton. J Clin Med. 2022 Feb 9;11(4):909.
3. Goyal G, Tazi A, Go RS, Rech KL, Picarsic JL, Vassallo R, Young JR, Cox CW, Van Laar J, Hermiston ML, Cao XX, Makras P, Kaltsas G, Haroche J, Collin M, McClain KL, Diamond EL, Girschikofsky M. International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults. Blood. 2022 Apr 28;139(17):2601-2621.
4. Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood. 2020 Apr 16;135(16):1319-1331.
5. Cantu MA, Lupo PJ, Bilgi M, Hicks MJ, Allen CE, McClain KL. Optimal therapy for adults with Langerhans cell histiocytosis bone lesions. PLoS One. 2012;7(8):e43257.
6. Aricò M, Girschikofsky M, Généreau T, Klersy C, McClain K, Grois N, Emile JF, Lukina E, De Juli E, Danesino C. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer. 2003 Nov;39(16):2341-8.
7. Makras P, Alexandraki KI, Chrousos GP, Grossman AB, Kaltsas GA. Endocrine manifestations in Langerhans cell histiocytosis. Trends Endocrinol Metab. 2007 Aug;18(6):252-7.
8. Yeh EA, Greenberg J, Abla O, Longoni G, Diamond E, Hermiston M, Tran B, Rodriguez-Galindo C, Allen CE, McClain KL; North American Consortium for Histiocytosis. Evaluation and treatment of Langerhans cell histiocytosis patients with central nervous system abnormalities: Current views and new vistas. Pediatr Blood Cancer. 2018 Jan;65(1).