Hemofilia A adquirida posparto, reporte de un caso
Postpartum acquired hemophilia A, a case report
Budde D, Finucci Curi B, Senor C, Suligoy J, Tardella M, Viollaz M.
Hospital Provincial de Rosario, Argentina.
residentesclinicamedicahpr@gmail.com
Palabras claves: hemofilia A,
postparto,
factor VIII,
puerperio
Keywords: hemophilia A,
postparthum,
factor VIII,
puerperium
Resumen
La hemofilia A adquirida (HAA) es una enfermedad hematológica infrecuente, caracterizada por la presencia de anticuerpos contra el factor VIII circulante. Se presenta en personas adultas y en la mitad de los casos en asociación a procesos autoinmunitarios, malignos (oncohematológicos y tumores sólidos), enfermedades dermatológicas, alergias y en mujeres durante embarazo, parto o puerperio. Las manifestaciones hemorrágicas son diversas, predominando el sangrado cutáneo espontáneo. El diagnóstico se realiza midiendo la actividad del factor VIII y la presencia de su inhibidor.
Reportamos el caso de una paciente de 36 años cursando 36° día de puerperio que se presenta con hematomas espontáneos, sangrado visceral y tiempos de coagulación prolongados. Se realizó diagnóstico de HAA posparto con buena evolución posterior al tratamiento inmunosupresor.
La HAA posparto presenta muy baja incidencia y, en consecuencia, existen pocos casos descriptos. Su retraso en el diagnóstico es un condicionante para el elevado porcentaje de mortalidad. De aquí el interés en la publicación de este caso.
Abstract
Acquired hemophilia A (HAA) is a rare hematological disease, characterized by the presence of antibodies against circulating factor VIII. It occurs in adults and in half of the cases in association with autoimmune, malignant processes (oncohematological and solid tumors), dermatological diseases, allergies and in women during pregnancy, childbirth or puerperium. Hemorrhagic manifestations are diverse, predominantly spontaneous skin bleeding. Diagnosis is accurate by measuring the activity of factor VIII and the presence of its inhibitor.
We present a case report of a 36-year-old patient in the early puerperium who presented with spontaneous hematomas, visceral bleeding and prolonged coagulation times. Postpartum HAA was diagnosed with good evolution after immunosuppressive treatment.
Postpartum HAA has a very low incidence and, consequently, there are few cases described. Its delay in diagnosis is a conditioning factor for the high percentage of mortality. Hence the interest in publishing this case.
Introducción
La hemofilia A adquirida (HAA) es una patología poco frecuente pero potencialmente grave, que suele cursar con manifestaciones hemorrágicas espontáneas y severas. Está definida como una enfermedad autoinmune, ya que las manifestaciones obedecen a la presencia de un autoanticuerpo inhibidor del factor VIII (IFVIII), que conduce a un trastorno de la coagulación. Puede ser idiopático o secundario a patologías agudas o crónicas, o a situaciones como embarazo y puerperio. Es importante la sospecha ante la presencia de sangrado espontáneo, ya que el retraso en el diagnóstico acrecienta la morbimortalidad. El tratamiento consiste en el uso de corticoides y complejo anti inhibidor, y el pronóstico depende de la causa subyacente y la gravedad de las manifestaciones(1).
Material y métodos
Presentamos un trabajo descriptivo y retrospectivo, por medio de revisión de historia clínica y revisión de la literatura. Cuenta con el aval de los Comités de Bioética y Docencia de nuestra institución. Este caso se rigió por las normas de la Declaración de Helsinki.
Caso clínico
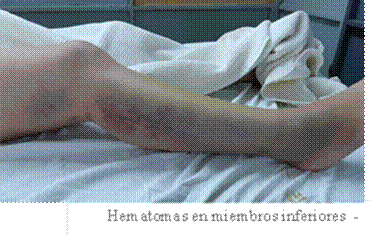
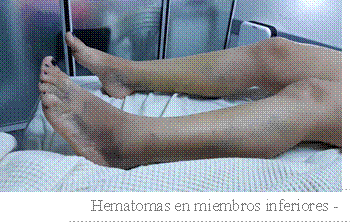
Paciente mujer de 36 años, que cursaba segundo mes de puerperio, con antecedentes de 2 gestas y 2 cesáreas. Consultó por un cuadro de 3 días de evolución de dolor lumbar derecho y hematuria, asociado a hematomas espontáneos en cara posterior de ambas piernas (Figuras 1 y 2). El cuadro se interpretó como pielonefritis aguda, por lo cual se comenzó con tratamiento antibiótico. Evolucionó con persistencia de los síntomas por lo cual se solicitó TAC de abdomen y pelvis con contraste endovenoso que evidenciaba dilatación pielo-ureteral derecha con ocupación de la pelvis renal y el uréter proximal por contenido hemático, con falta de eliminación del contraste (Figuras 3 y 4). Además se observaba litiasis renal bilateral. En el laboratorio de ingreso presentaba anemia normocítica, normocrómica y KPTT prolongado (Tabla 1) el cual no corrigió luego de la corrección con plasma. Se solicitó frotis de sangre periférica (FSP), estudios inmunológicos y dosaje de factor VIII (FVIII), e inhibidor del mismo (Tabla 2) realizando diagnóstico de hemofilia adquirida posparto.
Se inició tratamiento con pulso de metilprednisona (1 mg/kg/día por 3 días) asociado a complejo coagulante inhibidor - factor II, IX, X no activado y factor VIIa - FEIBA (50 U por kg -3000 UI- cada 12 hs) del cual realizó 7 dosis. Se realizó TAC de abdomen y pelvis de control con contraste al 5° día de internación donde se evidenció mejoría del sangrado visceral. Evolucionó afebril, sin nuevos signos de sangrados, con resolución de la hematuria y valores analíticos de tiempos de coagulación.
Al 9° día de internación se decide alta hospitalaria con prednisona 60 mg/día con controles por consultorio externo sin repetir episodios.

|
Fecha |
4 días previo al ingreso |
1 día |
3 día |
5 día |
7 día |
9 día |
|
Hematocrito / Hemoglobina |
30.8/10.7 |
29.2/10.1 |
30.6/10.6 |
|
29/10 |
35.2/12 |
|
VCM / HCM |
|
89/30 |
|
|
|
88/30 |
|
Glóbulos blancos (fórmula) |
8400 |
10100 69/0/0/17/11 |
8970 |
|
16470 |
17850 |
|
Plaquetas |
367000 |
389000 |
434.000 |
|
419000 |
406.000 |
|
Urea |
29/0.8 |
15/0.79 |
|
|
43/0.67 |
38/0.58 |
|
Ionograma |
140/4.8/100 |
|
|
|
142/3.5/102 |
136/3/95 |
|
LDH |
|
317 |
|
|
|
|
|
VES/<PCR |
70 |
45/4.2 |
|
|
56/0.3 |
39/0.3 |
|
Cpk |
|
120 |
|
|
|
|
|
TP-KPTT (seg.) |
14.8/58 |
11.4/60.2 |
9.6/59.7 |
10.2/43.6 |
10.5/36.9 |
10/36 |
|
Ca/P/Mg |
|
8.5/3.2/2.2 |
|
|
|
|
|
TGO/TGP/FAL |
|
13/12/79 0.6/0.2 |
|
|
|
|
|
Prot/Alb |
|
6.5/3.5 |
|
|
|
|
|
Colinesterasa |
|
6363 |
|
|
|
|
Laboratorio inmunológico:
● FAN: reactivo 1/40 - patente moteada
● Complemento C3: 179 (valor normal)
● Complemento C4: 18 (valor normal)
● Anticoagulante lúpico (coagulometría TVVR/AFL) 1.08 (negativo >1.2)
● Anticardiolipinas IgG: 9 (reactivo 20-80)
● Anticardiolipinas IgM: 26 (reactivo 20-80)
● Beta 2 glicoproteína I IgG: 13 (reactivo >20)
● Beta 2 glicoproteína I IgM: 9 (reactivo >20)
Actividad factor VIII: 3.8% (valor normal 50-150%)
Actividad factor IX: 78% (valor normal 50-200%)
Actividad factor XI: 90% (valor normal 65-130%)
Inhibidor factor VIII: 9 UB/ml (valor normal <0.5 Unidades Bethesda/ml)
Resultados
Una vez realizado el diagnóstico o ante una fuerte sospecha clínica, es importante iniciar con el tratamiento de manera urgente. Los objetivos del tratamiento terapéutico radican en controlar la hemorragia, disminuir los niveles de inhibidor de FVIII, aumentar los niveles del FVIII y tratamiento de la enfermedad subyacente.
Para el control de la hemorragia se inicia tratamiento con agentes hemostáticos. El tratamiento de primera línea es con fármacos con efecto bypaseante. Los fármacos con efecto bypass incluyen al factor VII activado (FVIIa) recombinante (Novoseven) y el concentrado de factores protrombínicos activados (Factor Eight Inhibitor Bypassing Agent, FEIBA, integrado por derivados del plasma que contiene factor II, IX, X, VIIa). A pesar de que se han asociado a mayor riesgo de efectos secundarios como infartos, trombosis o hemorragias, en el contexto de coagulación intravascular diseminada, sus beneficios son mayores, por lo cual integran la primera línea de tratamiento farmacológico. Por otro lado, los concentrados de FVIII pueden ser humanos, que son más efectivos en pacientes con títulos bajos (<5 BU) y porcinos, que son altamente efectivos incluso con niveles altos de inhibidores(1,5).
Otros agentes como la desmopresina (DDVAP) son de utilidad limitada en HAA. La desmopresina es un vasoconstrictor análogo sintético que libera el FVIII almacenado en combinación con factor von Willebrand (FVW) al sistema circulatorio e incrementa su nivel de 2 a 3 veces, lo que generalmente es suficiente para proporcionar hemostasia en caso de episodios hemorrágicos menores. Por dicha razón, son útiles en el tratamiento de personas con hemofilia leve y con títulos bajos de inhibidores (<2 BU/mL) y niveles de FVIII> 5 UI.
Asimismo, dentro de los agentes anti-fibrinolíticos se describe el ácido tranexámico y el ácido épsilon-aminocaproico (AECD). Ambos agentes actúan inhibiendo la activación del plasminógeno y de manera no competitiva la plasmina. El AECD se recomienda para las hemorragias bucales pero no para las hemorragias renales, como la de nuestra paciente(5).
En adición al tratamiento con agentes hemostático, el tratamiento inmunosupresor está recomendado en todo paciente con HAA para lo cual se utilizan fármacos como corticoides, ciclofosfamida, azatioprina e, incluso, de forma controversial, también se puede considerar el uso de inmunoglobulina G.
Considerando estudios recientes como UKHCDO, si bien no se demostraron diferencias significativas en cuanto al tiempo de remisión entre tratamiento con corticoides o corticoides + agentes citotóxicos, sí se puede destacar una mayor respuesta al tratamiento combinado. Sin embargo, el estudio EACH2, demostró que el uso de corticoides con ciclofosfamida (tratamiento combinado) vs rituximab o corticoides solos, generan menor tiempo de remisión(1).
En el caso de nuestra paciente, por presentar hemorragia visceral, se inició tratamiento con metilprednisona 1 mg/kg por 3 días y FEIBA 3.000 UI cada 12 hs endovenosas hasta la resolución del cuadro (control clínico y por tomografía).

*Tp y KPTT: segundos.
Discusión
La hemofilia adquirida A (HAA) es una enfermedad caracterizada por la presencia de anticuerpos contra FVIII de coagulación (inhibidores). El FVIII es una glicoproteína sintetizada a nivel del endotelio y que actúa como cofactor en la cascada de la coagulación. El mismo es independiente de vitamina K y circula de forma inactivada junto con el FVW; se activa al entrar en contacto con lesión endotelial y activan, junto con el factor IXa, calcio y fosfolípidos, al factor X.
Su incidencia es de 1,5 casos por millón por año y se presenta principalmente en adultos (edad media 64 - 68 años), pero puede estar asociada a mujeres jóvenes en contexto de enfermedades autoinmunitarias, embarazo y puerperio(1).
La HAA es una enfermedad autoinmune donde se generan IgG contra el FVIII de la coagulación como consecuencia de la interacción de factores genéticos y ambientales. Se ha observado que alrededor de la mitad de los pacientes con HAA presenta un grupo heterogéneo de procesos patológicos y fisiológicos asociados que incluyen, entre otros, enfermedades autoinmunes (9.4-17%) y malignas (6.4-18.4%), embarazo, parto y puerperio. Aproximadamente en la mitad de los pacientes la HAA se define como idiopática, ya que no se asocia a ningún proceso(1,3).
La HAA posparto es una entidad rara (100 casos descriptos en la literatura) y representa el 7-11 % de las HAA. Predomina en mujeres primigestas, pero se ha descripto en multíparas, como en nuestro caso(3).
Los síntomas comienzan normalmente en los primeros 3 meses tras el parto, como es el caso de nuestra paciente quien inicia con signos y síntomas a los 2 meses de su segunda cesárea, pero pueden aparecer hasta 12 meses después. La HAA posparto suele presentarse con metrorragia sin causa obstétrica aparente (y en ausencia de historia clínica hematológica previa) pero pueden presentarse con hematomas en tejidos blandos y viscerales. Clínicamente nuestra paciente presentó hematomas espontáneos en tejido subcutáneo (sitio más frecuente de sangrado > 80%) y hematuria secundaria a sangrado visceral en pelvis renal.
El diagnóstico de HAA se realiza mediante pruebas de laboratorio. Un tiempo parcial de tromboplastina activada prolongado (KPPT) con un tiempo de protrombina (TP) normal indica un defecto en la vía intrínseca de la coagulación y siempre debe ser estudiado (Anexo 3). En primer lugar se debe realizar una prueba con plasma normal, la cual modifica el KPTT si el defecto es debido al déficit del factor VIII, mientras que el KPTT no se modifica en presencia de un inhibidor del mismo(4).
Posterior a esta prueba, como lo hemos realizado con nuestra paciente, se debe realizar detección y titulación del inhibidor FVIII. La detección se realiza mediante dos pruebas: el índice de Rosner y potenciación con tiempo y temperatura. El IR (RI = (mezcla-N)/P × 100) se expresa en porcentajes: si el valor obtenido es menor al 10% significa que hay ausencia de inhibidor, pero si el IR es mayor al 10% implica presencia del mismo, como lo fue en el caso de nuestra paciente. La prueba de potenciación con tiempo y temperatura implica exponer al plasma a 37º C, en un tiempo determinado, evento que potencia al inhibidor y prolonga aún más el KPTT en presencia del mismo.
La titulación y el diagnóstico definitivo se realiza cuantificando el inhibidor del FVIII mediante el ensayo Bethesda (BA), ensayo Nijmegen Bethesda (NBA), o en algunos laboratorios por un ensayo inmunoabsorbente ligado a enzimas (ELISA) con anti-FVIII anticuerpo.
En nuestro caso, frente a valores analíticos de KPTT prolongados, se realizó la prueba de modificación con plasma normal, no obteniendo resultados favorables. Por dicha razón, y frente a la alta sospecha clínica, se solicitó dosaje de inhibidor de FVIII, evidenciando un índice de Rosner mayor a 10% y se realizó mediante el ensayo Bethesda la cuantificación de su inhibidor; corrigiendo el KPTT y negativizando el inhibidor luego del tratamiento con complejo coagulante.
Por último, es importante destacar que la HAA posparto es una enfermedad de baja incidencia, con buen pronóstico a largo plazo, tasa de recurrencia cercana a la nulidad y, principalmente, que la posibilidad de remisión espontánea de los inhibidores es alta (cercana a un 100%). Esto es diferente para HAA de otras etiologías como, por ejemplo, asociado a patologías autoinmunes y oncológicas, casos en los cuales la resolución depende de los tratamientos de las enfermedades de base. Sin embargo, el tratamiento inmunosupresor y con agentes hemostáticos se asocia a menor tiempo de remisión y menor riesgo de hemorragias que comprometerían la vida del paciente, por lo cual, se debe iniciar precozmente el tratamiento adecuado frente a sospecha clínica y/o diagnóstico de la enfermedad.
Bibliografía
1. Kruse-Jarres R, Kempton CL, Baudo F y col. Acquired hemophilia A: updated review of evidence and treatment guidance. Wiley AJH. 2017;92:695-705.
2. Arbesú G, Dávoli M, Elhelou L y col. Hemofilia. Guía de diagnóstico y tratamiento. Sociedad Argentina de Hematología. 2017;167-177.
3. Del Valle Rubido C, Fernández Muñoz L, Cano Cuetos A, Solano Calvo JA, Pascual T, Zapico Coñi A. Hemofilia A adquirida posparto: guía de diagnóstico y tratamiento a raíz de un caso clínico. Prog Obstet Ginecol. Elsevier Doyma. 2014;57(3);135-139.
4. Philippa D, Helen A. Acquired haemophilia A: a rare cause of postpartum haemorrhage. NZMJ. 2018;131:111-115.
5. Hemophilia of Georgia. Federación Mundial de Hemofilia. Protocolos para el tratamiento de la hemofilia y de la enfermedad de von Willebrand. Tercera edición. 2008; Nº 14.