Valor pronóstico de las anomalías genéticas en mieloma múltiple
Prognostic value of genetic abnormalities in multiple myeloma
Flavia Stella1,2, Leticia Giselle Guash1, Estela Pedrazzini1,3, Irma Slavutsky1
1. Laboratorio de Genética de Neoplasias Linfoides, Instituto de Medicina Experimental, CONICET-Academia Nacional de Medicina, Buenos Aires, Argentina
2. Área de Genética, Servicio de Anatomía Patológica, Hospital Posadas, Buenos Aires, Argentina
3. Departamento Ciencias Básicas y Experimentales, Universidad del Noroeste de la Provincia de Buenos Aires (UNNOBA), Buenos Aires, Argentina
islavutsky@hematologia.anm.edu.ar
Palabras clave: mieloma múltiple,
citogenética,
FISH.
Keywords: multiple myeloma,
cytogenetics,
FISH.
Resumen
El mieloma múltiple (MM) es una neoplasia B post centro germinal caracterizada por la proliferación clonal de células plasmáticas en la médula ósea y la detección de una inmunoglobulina monoclonal en suero u orina, la proteína M. Se origina a partir de un proceso de transformación de múltiples pasos con acumulación progresiva de eventos genéticos que favorecen la proliferación y expansión del clon maligno. A nivel citogenético se observan anomalías primarias, directamente relacionadas con la patogénesis de la enfermedad, y secundarias, que incluyen ganancias y pérdidas de material genético, que aportan información de valor pronóstico adicional. Las anomalías primarias permiten dividir a los pacientes en dos grandes grupos: hiperdiploides, con ganancia de cromosomas, considerados de buen pronóstico, y no-hiperdiploides, con número modal variable, asociados a mala evolución clínica. Las anomalías secundarias incluyen: deleción 13q14/monosomía del cromosoma 13, deleción 17p13, alteraciones del cromosoma 1, específicamente ganancia/amplificación del brazo largo (1q21) y deleciones del brazo corto (1p), así como también rearreglos del gen MYC (8q24). Un nuevo subgrupo lo constituyen los MM doble hit que incluyen pacientes con: a) inactivación bialélica de TP53 (deleción en un alelo y mutación en el otro) y, b) estadio clínico ISS III con amplificación de 1q21 (≥4 copias), asociados a muy mal pronóstico. Una anomalía de reciente descripción es la inactivación bialélica del gen BCMA (B cell maturation antigen) (16p13.13), que constituye un mecanismo de recaída/resistencia al tratamiento anti-BCMA CAR T-cells, siendo importante su detección en pacientes pasibles de ser incorporados a estos esquemas terapéuticos. Sin duda, la profundización de la caracterización biológica del MM resulta de fundamental importancia en el marco de una medicina traslacional, contribuyendo a un mejor diagnóstico y/o pronóstico, y aportando información para nuevos abordajes terapéuticos.
Summary
Multiple myeloma (MM) is a clonal B cell neoplasia characterized by the accumulation of malignant plasma cells within the bone marrow and the presence of a monoclonal immunoglobulin in the serum and/or urine. MM originates from a multi-step transformation process with progressive accumulations of genetic events that favor the proliferation and expansion of the malignant clone. The disease is characterized by the presence of multiple structural and numerical genetic alterations with different prognostic significance. Chromosome alterations can be divided in primary, related to the pathogenesis of the disease, and secondary, that provide additional prognostic information. Primary alterations permit classify patients in two groups: hyperdiploid, with chromosome gains, considered of favorable prognosis, and non-hyperdiploid, with variable modal number and associated to poor clinical evolution. The secondary abnormalities include: 13q14 deletion/monosomy of chromosome 13, deletion of 17p13, chromosome 1 alterations, particularly gains/amplifications of 1q21 and 1p deletions, and structural rearrangements of MYC gen (8q24). In addition, there is a new subgroup named double hit MM that includes patients with: a) biallelic inactivation of TP53 gene (deletion in one allele and mutation in the other) and, b) clinical stage ISS III and 1q21 amplification (≥4 copies), associated to very poor prognosis. The biallelic inactivation of the BCMA (B cell maturation antigen) gene (16p13.13) is a new alteration, associated to relapse/resistance to the anti-BCMA CAR T-cells treatment, which detection is of relevance, particularly in those patients eligible for these therapeutic protocols. The deepening of the biologic characterization of MM results of importance in the context of translational medicine, contributing to a better diagnosis and/or prognosis, and providing information for new therapeutic approaches.
Introducción
El MM es la segunda neoplasia hematológica en frecuencia en adultos del mundo occidental(1). Afecta más comúnmente a individuos con edad media al diagnóstico de 65-70 años; es más frecuente en hombres que en mujeres, y presenta diferencias en su incidencia entre regiones geográficas y grupos étnicos(2). Es una enfermedad heterogénea en cuanto a su presentación clínica, respuesta a la terapia y tiempo de sobrevida (SV)(3). A nivel genético es una patología compleja que sufre un proceso de transformación de múltiples pasos con acumulación progresiva de eventos que favorecen la proliferación y expansión del clon maligno(3-6). El estudio de las alteraciones genéticas ha permitido la definición de subgrupos específicos, y provisto las bases para la identificación de genes involucrados en la iniciación y progresión de esta entidad, siendo primordiales al momento del diagnóstico, en la progresión/recaída de la enfermedad, así como en la evaluación de la respuesta al tratamiento(4,7).
El análisis citogenético presenta limitaciones debido al bajo índice de proliferación de las células plasmáticas (CP), permitiendo detectar alteraciones cromosómicas en el 30-40% de los casos(4). Un trabajo reciente(8) muestra el valor pronóstico independiente del cariotipo convencional en la detección de pacientes de alto riesgo, rescatando la importancia de esta metodología en el estudio del MM. Su complementación con la técnica de FISH (Fluorescence in situ hybridization) permitió aumentar considerablemente la detección de alteraciones, llegando aproximadamente al 80% de los casos, siendo de importancia dada la presencia de numerosas anomalías crípticas.
Las alteraciones cromosómicas incluyen anomalías primarias, directamente relacionadas con la patogénesis de la enfermedad, y secundarias, particularmente ganancias y pérdidas de material genético, que aportan información pronóstica adicional(7,9) y que, en conjunto, han permitido establecer diferentes grupos de riesgo. Las anomalías primarias permiten dividir a los MM en dos grandes grupos: hiperdiploides (42-45% de los casos) y no-hiperdiploides (35-40%). Asimismo, se observa un 10-15% de pacientes con alteraciones combinadas (hiperdiploidía y t14q32) y un 5-10% que presenta anomalías variables, cuyo comportamiento clínico es heterogéneo(10). Recientemente, se ha identificado un nuevo subgrupo con cariotipo tetraploide (4n) de muy mal pronóstico (6% de los MM al diagnóstico), asociados a mayor infiltración de CP en la médula ósea (MO) y aumento de anomalías de alto riesgo por FISH(11). Asimismo, el MM se caracteriza por una alta heterogeneidad clonal, aún en estadios iniciales de la enfermedad, con diferentes patrones que van desde la estabilidad clonal a la presencia de multiclonalidad(5,6,12), resultando importante su evaluación dada sus implicancias a nivel del tratamiento.
Anomalías primarias
Los MM hiperdiploides presentan entre 48 y 74 cromosomas y se caracterizan por mostrar múltiples trisomías, preferencialmente de los cromosomas impares 3, 5, 9, 11, 15, 19 y/o 21(4,12) (Figura 1a). Se manifiestan en individuos de mayor edad y se asocian a lesiones óseas y pronóstico favorable, no siendo claro el mecanismo subyacente en su desarrollo. No todas las trisomías tienen la misma frecuencia, siendo la más común la trisomía 9 seguida por los cromosomas 15 y 19, y, en menor frecuencia, la trisomía 17(13). Kumar y col.(14) observaron una atenuación del impacto adverso de las alteraciones de alto riesgo por parte de las trisomías, mientras que Chretien y col.(13) detectaron que las trisomías 3 y 5 mejoran significativamente el pronóstico de los pacientes, presentando un rol protector en casos con t(4;14) y del(17)(p13), en tanto que la trisomía 21 lo empeora. Estos datos llevaron a la revisión y modificación del riesgo citogenético determinando el desarrollo de un nuevo índice pronóstico que incluye a las trisomías(15,16). También se observó mejor pronóstico en los pacientes con hiperdiploidías superiores a 50 cromosomas respecto de aquellos con hiperdiploidías entre 47-50 cromosomas. Otros trabajos(17,18) indican que la presencia de alteraciones de alto riesgo en pacientes con MM hiperdiploide impacta negativamente en el pronóstico de los mismos.
Figura 1. Cariotipos de pacientes con MM. a) cariotipo hiperdiploide; b) cariotipo no-hiperdiploide.
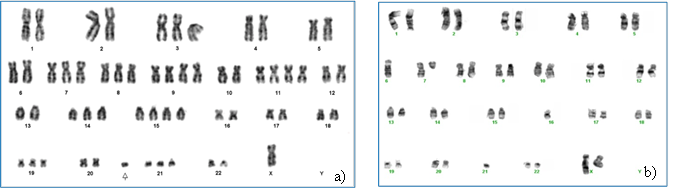
Por su parte, los MM no-hiperdiploides presentan un número modal variable, desde hipodiploide hasta pseudotetraploide (Figura 1b). Constituyen un grupo heterogéneo, con numerosos subtipos moleculares asociados a translocaciones recurrentes que involucran al gen de la cadena pesada de las inmunoglobulinas (IGH), ubicado en 14q32, y cinco diferentes oncogenes: FGFR3-MMSET (4p16), CCND3 (6p21), CCND1 (11q13), MAF (16q23) y MAFB (20q12), con implicancias pronósticas(10,19) (Tabla 1). En menor frecuencia se observan otras translocaciones del cromosoma 14, entre ellas la t(6;14)(p25;q32) que involucra al gen IRF4 (Interferon Regulatory Factor 4). Todas ellas determinan la desregulación de ciclinas D, que lleva a la activación del ciclo celular y aporta ventajas selectivas a los subclones que las presentan(20).
Tabla 1. Subgrupos de alteraciones citogenéticas en MM no-hiperdiploide
|
Subgrupo |
Genes involucrados |
Frecuencia (%) |
|
Rearreglos de ciclinas |
|
|
|
t(11,14)(q13;q32) |
CCND1/IGH |
15-20 |
|
t(6,14)(p21;q32) |
CCND3/IGH |
3-6 |
|
t(12;14)(p13;q32) |
CCND2/IGH |
<1 |
|
|
|
|
|
t(4;14)(p16;q32) |
FGFR3-MMSET/IGH |
6-15 |
|
|
|
|
|
Rearreglos de MAF |
|
|
|
t(14;16)(q32;q23) |
IGH/MAF |
4-5 |
|
t(14;20)(q32;q11) |
IGH/MAFB |
1-2 |
|
t(8;14)(q24.3;q32) |
MAFA/IGH |
<1 |
CCND: ciclina D; IGH: immunoglobulin heavy chain; FGFR3: fibroblast growth
factor receptor 3; MMSET: multiple myeloma SET domain; MAF:
musculoaponeurotic fibrosarcoma oncogene
En lo que respecta a las translocaciones que involucran a los genes de ciclinas (CCND), la más frecuente es la t(11;14)(q13;q32), observada en el 15-20% de los pacientes al diagnóstico, que determina la sobreexpresión de CCND1(4,10). Puede detectarse en MGUS (monoclonal gammopathy of undetermined significance) y presenta alta incidencia en la leucemia de células plasmáticas (LCP), tanto primaria como secundaria (50-60% de los casos)(21). Tiene características biológicas y clínicas distintivas, está asociada con enfermedad hipo-secretoria o no-secretoria, IgD o IgM, morfología linfoplasmocítica o de pequeñas CP maduras, cadena liviana lambda, expresión aumentada de CD20, asociada a mejor SV global y libre de progresión, y pérdida de expresión de CD56(21-25). Estos pacientes muestran aumento de la expresión de la proteína antiapoptótica BCL-2 y disminución de las proapoptóticas MCL-1/BCL-XL(26), lo que los hace susceptibles al tratamiento con venetoclax, un inhibidor de BCL-2(27). Si bien originalmente esta translocación ha sido asociada con un riesgo estándar, estudios recientes muestran peor evolución respecto de los casos con cariotipo y FISH normal y mejor respuesta que los pacientes con alteraciones de alto riesgo(28,29). Las otras dos translocaciones que involucran genes de ciclinas son t(6;14)(p21;q32) (CCND3) y t(12;14)(p13;q32) (CCND2), de muy baja frecuencia (2% y <1%, respectivamente), y con escasa información clínica(20).
La t(4;14)(p16.3;q32) es la segunda translocación recíproca en frecuencia en los pacientes con MM, observada en aproximadamente el 15% de los casos(30), asociada a IgA y cadena liviana lambda. Es una translocación críptica que debe ser evaluada mediante FISH. Determina la yuxtaposición de dos oncogenes: MMSET (multiple myeloma SET domain) y FGFR3 (fibroblast growth factor receptor 3), ubicados en 4p16.3, con el gen IGH. MMSET permanece en el derivado 4 y se encuentra sobreexpresado en el total de casos, en tanto que la expresión del receptor de tirosina quinasa FGFR3 se manifiesta en el 75% de los pacientes debido a la pérdida del derivado 14 (25% de los casos con esta translocación)(31-33). MMSET codifica para una histona metiltransferasa con un rol central en la patogénesis del MM, asociada a progresión tumoral e inestabilidad genómica(33-35). Por el contrario, diferentes estudios demostraron que la expresión de FGFR3 no tiene impacto significativo en la sobrevida de los pacientes(32,34), en tanto que un análisis reciente revela que los casos sin t(4;14) pero con un perfil de expresión génica MMSET tienen un pronóstico similar a aquéllos que presentan la translocación(35). Los pacientes con esta anomalía constituyen un grupo heterogéneo en cuanto a su evolución clínica(37), lo cual estaría relacionado a la presencia de distintos transcriptos de MMSET(32,33), que impactarían de manera diferencial en la evolución clínica de la enfermedad(38), así como a la coexistencia de otras alteraciones capaces de modular el pronóstico de esta translocación, como se evidencia en el nuevo índice pronóstico recientemente establecido(15,16). Particularmente, la t(4;14) con coexistencia de alteraciones del cromosoma 1 y/o trisomía 21 presenta alto riesgo citogenético (29% de los casos)(39), en tanto que aquéllos con t(4;14) y trisomía 5 se asocian a un mejor pronóstico.
Las translocaciones que involucran a los genes MAF (Musculoaponeurotic fibrosarcoma) son menos frecuentes, están asociadas al isotipo IgA y se encuentran relacionadas a pronóstico adverso(10). Ellas incluyen la t(14;16)(q32;q23) (5% de los casos) y la t(14;20)(q32;q11) (2%) que yuxtaponen IGH con los oncogenes MAF y MAFB, respectivamente(4,10); ambas son translocaciones crípticas que deben detectarse mediante FISH. Dichos genes son factores de transcripción de la familia bZIP (Basic leucine zipper), que proviene de la superfamilia AP-1, y tiene funciones de transactivación(40-42). Estas translocaciones determinan el aumento de expresión de los genes MAF, que lleva a una sobre-expresión de CCND2, promoviendo la proliferación celular y afectando la regulación de la transición G1/S del ciclo celular. Asimismo, generan aumento de la expresión de la integrina β7 que promueve la adhesión de las células de MM con las células estromales de la MO, fenómeno que se asocia con el aumento de la secreción de VEGF (Vascular Endothelial Growth Factor), importante en la SV de las células mielomatosas, y relacionado a resistencia a la apoptosis(43). Estudios de microarrays muestran un perfil de expresión génica similar para los genes MAF y MAFB, sustentando un camino de señalización común para ambos(44). Los puntos de ruptura de la t(14;16) se producen en el último intrón del gen supresor tumoral WWOX (WW domain containing oxidoreductase), centromérico a MAF, donde se encuentra el sitio frágil FRA16D, generando su disrupción y activación por parte del enhancer de IGH(45,46). Estos pacientes presentan baja expresión de CD56, aumento de la expresión de CD20 y alteraciones cromosómicas en el cariotipo(47). Un trabajo reciente(48) no observa diferencias en la media de SV entre los pacientes portadores de la t(14;16) y aquéllos que no la tenían, señalando un interrogante sobre su significado pronóstico. Con respecto a la t(14,20), resulta interesante mencionar que su presencia en MGUS y MM indolente se asocia a enfermedad estable, por lo que se sugiere que la misma no sería responsable del mal pronóstico observado en los pacientes con MM, requiriendo eventos adicionales para la progresión de la enfermedad (10,19,49).
Alteraciones secundarias
Las mismas incluyen: deleción 13q14, del17p13, ganancia/amplificación de 1q21 y del1p, y translocaciones que involucran al oncogén MYC (Myelocytomatosis) (8q24)(1,14,22).
La del13q14 es una de las anomalías más comunes (45-50% de los casos)(4); involucra al gen supresor de tumor RB1 (Retinoblastoma), regulador negativo del ciclo celular. Se la detecta también en MGUS, lo que sugeriría un rol primario en la oncogénesis temprana(50). Aproximadamente el 85% de los pacientes presenta monosomía 13, mientras que en el 15% restante se producen deleciones intersticiales(51). Presenta un alto nivel de asociación con las translocaciones que involucran IGH (84%), llegando al 90% de los casos que tienen la t(4;14)(p16;q32)(4). Si bien durante mucho tiempo se la consideró un marcador de pronóstico adverso, actualmente se sabe que su detección indica la presencia de hipodiploidía o translocaciones IGH, resultando un marcador molecular de MM no-hiperdiploide(19). Su detección en metafase se asocia a mayor proliferación y masa tumoral(51,52). Un trabajo reciente(53) encuentra efectos diferentes sobre la SV global y la progresión al analizar por separado los pacientes con del(13q) y aquéllos con monosomía 13, evidenciando efecto protector para la deleción y adverso para la monosomía, en tanto que la presencia de inactivación bialélica de RB1 se relacionó a peor evolución clínica, con valor pronóstico independiente en la recaída de la enfermedad(54).
La del17p13, lugar donde mapea el gen supresor de tumor TP53 (Tumor supresor P53), es el factor de pronóstico adverso más importante en MM(4,12,55,56), asociada a alto riesgo en el R-ISS (Revised International Staging System)(57). Se la observa en aproximadamente el 10% de los pacientes al diagnóstico, llegando al 80% en los últimos estadíos de la enfermedad, y se vincula con resistencia al tratamiento. El gen TP53 funciona como un regulador transcripcional que participa en el control del ciclo celular, reparación y respuesta al daño del ADN, y promoción de la apoptosis, siendo uno de los genes más frecuentemente mutados en cánceres humanos. Su pérdida se relaciona a enfermedad agresiva, corta SV, enfermedad extramedular, hipercalcemia y compromiso del sistema nervioso central(10,12,58,59). Se presenta en la mayoría de los casos con LCP tanto primaria como secundaria y es muy infrecuente en MGUS(60).
Las anomalías del cromosoma 1, específicamente ganancias de 1q21, presentes en el 35-40% de los pacientes(61,62) y pérdidas de 1p observadas en el 30% de los casos(63,64), se encuentran entre las alteraciones estructurales más frecuentes en el MM, comúnmente asociadas con progresión de la enfermedad(4,9,10,12). En cuanto a la ganancia/amplificación de 1q, el gen más involucrado es CKS1B (CDC28 protein kinase regulatory subunit 1B), que mapea a nivel de 1q21.3; codifica para un regulador positivo del ciclo celular que activa las quinasas dependientes de ciclinas, promoviendo la proliferación celular(65). La sobreexpresión de CKS1B se encuentra asociada a alto nivel de proliferación y pronóstico adverso(61,66,67). El análisis del nivel de expresión de CKS1B y del número de copias mostró mayor valor pronóstico para este último(66.67), en tanto que Stella y col.(66) detectaron mayor expresión de CKS1B en MM respecto de MGUS, sugiriendo un rol en la progresión de MGUS a MM. Las alteraciones de 1q21 muestran una fuerte asociación con otras anomalías recurrentes en MM como translocaciones de IGH, del13q14 y del17p13, cuya presencia claramente empeora el pronóstico de los pacientes(68). Simultáneamente, las alteraciones de 1q han sido relacionadas con la presencia de translocaciones jumping y un mayor nivel de inestabilidad genómica(69). Al presente hay otros genes de interés en la región, como ADAR1 (Adenosine Deaminase RNA Specific) (1q21.3), que codifica para una enzima responsable de la edición del ARN, e IL6R (Interleukina 6 membrane receptor), subunidad del receptor de transmembrana de IL6, que confieren oncogenicidad a través de la activación de la vía de señalización JAK/STAT3(70-72). Particularmente, el aumento de expresión de ADAR1 contribuye a la patogénesis del MM y estimularía la progresión y resistencia a la terapia(72).
En cuanto a la del1p, encontramos dos regiones de importancia, 1p12 y 1p32.3. La primera contiene al gen supresor tumoral FAM46C (Family With Sequence Similarity 46 Member C), cuya función es de importancia en la síntesis proteica(73), también asociado a mala evolución clínica(64). El punto de ruptura 1p32.3 se encuentra afectado con mayor frecuencia, siendo CDKN2C (Cyclin dependent Kinase Inhibitor 2C) el gen más involucrado, que participa en la regulación negativa del ciclo celular(74), cuya desregulación se vincula con significado clínico adverso(75,76). Si bien no es un evento frecuente, diferentes autores han detectado deleciones homocigotas de los genes involucrados en 1p, asociadas a mala evolución clínica(64,77). El análisis conjunto de estas anomalías permitió establecer grupos de riesgo citogenético (Tabla 2), algunos de los cuales se encuentran en revisión.
Tabla 2. Grupos de riesgo citogenético
|
Riesgo alto (20-25%) |
|
t(4;14)(p16;q32) (FISH) |
|
t(14;16)(q32;q23) (FISH) |
|
t(14;20)(q32;q11) (FISH) |
|
del(17)(p13) (FISH) |
|
del(1p) (citogenética o FISH) |
|
Ganancia de 1q (citogenética o FISH) |
|
Cariotipo complejo Doble hit |
|
|
|
Riesgo estándar (75-80%) |
|
Hiperdiploidía |
|
t(11;14)(q13;q32) (citogenética o FISH) |
|
t(6;14)(p21;q32) (citogenética o FISH)
|
Modificado de Rajkumar(10)
Además, los pacientes con MM pueden presentar rearreglos del gen MYC en, aproximadamente, el 15% de los casos recién diagnosticados, llegando al 45% durante la progresión(78). La sobrexpresión de este gen se asocia con formas más agresivas de la enfermedad, LCP y enfermedad extramedular(79-81). El mismo tiene un rol central en el crecimiento y proliferación celular, replicación del ADN, síntesis proteica y metabolismo energético(82). El mecanismo de activación se origina principalmente a través de translocaciones que involucran a los genes de las cadenas pesada y livianas de las inmunoglobulinas (50% de los casos) así como a FAM46C, FOXO3, BMP6, entre otros genes, en el 50% restante(83). Diferentes estudios mostraron su activación durante la transición de MGUS a MM, indicando su implicancia en la progresión de la enfermedad, y sustentando la dependencia de las células de MM a dicho oncogén para su supervivencia(78,80,84). Su presencia en pacientes con cariotipos hiperdiploides constituye un factor de pronóstico adverso(18). Un trabajo reciente de nuestro laboratorio(85) muestra rearreglos de MYC en el 11,5% de los pacientes, significativamente asociados a la presencia de falla renal.
Recientemente se detectaron nuevas anomalías de valor pronóstico. Entre ellas encontramos la deleción de la región variable del gen IGH y la amplificación de su región constante evaluadas por FISH, asociadas a peor pronóstico y corta SV libre de progresión(86) y la inactivación bialélica del gen BCMA (B cell maturation antigen), ubicado en 16p13.13. Éste último pertenece a la superfamilia del receptor de TNF (Tumor necrosis factor) (TNFRSF17: TNF Receptor Superfamily Member 17), es importante para el desarrollo de las células B, se expresa preferencialmente en linfocitos B maduros y resulta esencial para la supervivencia de las CP(87). La del16p se observa en el 6,7% de los pacientes con MM, presentando co-ocurrencia con la del17p en el mismo clon en el 77% de los casos. A su vez, el 36% de los pacientes con del17p muestran del16p(88,89). La sobreexpresión y activación de BCMA se asocian a progresión del MM, constituyendo un blanco terapéutico atractivo, ya sea como anticuerpo biespecífico, anticuerpo fármaco-conjugado o como anti-BCMA CAR T-cells(87). La inactivación bialélica de BCMA constituye un mecanismo de recaída/resistencia a este último tratamiento(88,89), siendo importante poder detectar esta anomalía en pacientes pasibles de ser incorporados a estos esquemas terapéuticos.
Otro aspecto de importancia es la presencia de cariotipos complejos (tres o más alteraciones cromosómicas), originados a partir de la acumulación de cambios secuenciales capaces de desregular mecanismos genéticos relacionados al desarrollo y/o progresión de la enfermedad(8,9,90). Los mismos se encuentran significativamente asociados a pobre evolución clínica, constituyendo un factor pronóstico independiente y resaltando el valor del estudio citogenético en el MM.
MM doble hit
Recientemente Walker y col(91) identificaron un nuevo subgrupo de pacientes con MM de alto riesgo y enfermedad muy agresiva, con características biológicas y clínicas específicas y muy corta SV libre de progresión y global a pesar de los nuevos tratamientos, denominado MM doble hit. Este subgrupo corresponde a aproximadamente el 6% del total de los casos e incluye pacientes con: a) inactivación bialélica de TP53 (deleción en un alelo y mutación en el otro) y, b) estadio clínico ISS III con amplificación de CKS1B (≥4 copias). Más recientemente, se incluyó entre los MM doble hit a los pacientes con cariotipo hiperhaploide, una alteración numérica rara, que presenta entre 24 y 34 cromosomas, corresponde al 0,25% de los casos con MM al diagnóstico(92), y afecta mayormente a individuos jóvenes(93). La presencia de cariotipos hiperhaploides es un evento muy poco frecuente en MM(94,95), pero es observado en leucemias agudas y algunos tumores sólidos(96). En MM, el clon hiperhaploide se caracteriza por presentar monosomía de numerosos cromosomas en tanto que los cromosomas impares, asociados a trisomías en los casos hiperdiploides, permanecen disómicos. Estos pacientes muestran con frecuencia monosomía del cromosoma 17, usualmente asociada a mutación de TP53 en el alelo restante, llevando a inactivación bialélica de dicho gen, quedando por lo tanto incluidos en el grupo de MM doble hit de muy alto riesgo(91). Si bien no se conoce exactamente el mecanismo de origen de estos cariotipos, diferentes autores sugieren, en base a estudios secuenciales, que el cariotipo hiperhaploide se originaría por la pérdida de cromosomas a partir de un cariotipo diploide, como consecuencia de defectos en el huso mitótico y/o en los centrosomas que llevarían a una segregación anormal(91,92).
Estos estudios sustentan la importancia de las alteraciones genéticas en la compleja heterogeneidad que caracteriza al MM y su implicancia en la presentación clínica y respuesta al tratamiento. Dicha complejidad aumenta considerablemente durante la progresión de la enfermedad con la adquisición de anomalías citogenéticas secundarias y mutaciones que se suman a las alteraciones primarias de cada subtipo, modificando el fenotipo clínico. Sin duda, la profundización de la caracterización biológica del MM resulta de fundamental importancia en el marco de una medicina adaptada al riesgo, contribuyendo a un mejor diagnóstico y/o pronóstico, y aportando información para nuevos abordajes terapéuticos.
Agradecimientos
El presente trabajo se efectuó con subsidios de Proyectos de Investigación Interinstitucionales de la Universidad de Morón y del Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET).
Bibliografía
1. Seer Cancer Statistics Review. Seer Cancer Statistics Review. Bethesda, MD: National Cancer Institute. 2018. Available online at: https://seer.cancer.gov/csr/1975_2015/
2. Colunga-Pedraza PR, Gómez-Cruz GB, Colunga-Pedraza JE, Ruiz-Argüelles GJ. Geographic hematology: Some observations in Mexico. Acta Haematol. 2018;140:114-20.
3. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364:1046-60.
4. Fonseca R, Bergsagel PL, Drach J y col. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23:2210- 21.
5. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12:335-48.
6. Corre J, Munshi N, Avet-Loiseau H. Genetics of multiple myeloma: another heterogeneity level? Blood. 2015;125:1870-6.
7. Kumar SK, Rajkumar SV. The multiple myelomas - current concepts in cytogenetic classification and therapy. Nat Rev Clin Oncol. 2018;15:409-21.
8. Soekojo CY, Wang C-M, Chen Y y col. Role of conventional karyotyping in multiple myeloma in the era of modern treatment and FISH analysis. Clin Lymph Myeloma. Leuk 2019;19:e470-7.
9. Stella F, Pedrazzini E, Agazzoni M, Ballester O, Slavutsky I. Cytogenetic alterations in multiple myeloma: Prognostic significance and the choice of frontline therapy. Cancer. Invest 2015;27:1-9.
10. Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, risk-stratification, and management. Am J Hematol. 2020;95:548-67.
11. Sidana S, Jevremovic D, Ketterling RP y col. Tetraploidy is associated with poor prognosis at diagnosis in multiple myeloma. Am J Hematol. 2019;94:e117-20.
12. Manier S, Salem KZ, Park J y col. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14:100-3.
13. Chretien ML, Corre J, Lauwers-Cances V y col. Understanding the role of hyperdiploidy in myeloma prognosis: which trisomies really matter? Blood. 2015;126:2713-9.
14. Kumar S, Fonseca R, Ketterling RP y col. Trisomies in multiple myeloma: impact on survival in patients with high-risk cytogenetics. Blood. 2012;119:2100-5.
15. Perrot A, Lauwers-Cances V, Tournay E y col. Development and validation of a cytogenetic prognostic index predicting survival in multiple myeloma. J Clin Oncol. 2019; 37:1657-65.
Blood. 2021;137:16-9.
17. Pawlyn C, Melchor L, Murison A y col. Coexistent hyperdiploidy does not abrogate poor prognosis in myeloma with adverse cytogenetics and may precede IGH translocations. Blood. 2015;125:831-40.
18. Weinhold N, Kirn D, Seckinger A y col. Concomitant gain of 1q21 and MYC translocation define a poor prognostic subgroup of hyperdiploid multiple myeloma. Haematologica. 2016;101:e116-9.
19. Rajan AM, Rajkumar SV. Interpretation of cytogenetic results in multiple myeloma for clinical practice. Blood Cancer J. 2015;5:e365.
20. Kuehl WM, Bergsagel PL. Molecular Pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest. 2012;122:3456-63.
21. An G, Xu Y, Shi L y col. t(11;14) Multiple myeloma: A subtype associated with distinct immunological features, immunophenotypic characteristics but divergent outcome. Leuk Res. 2013;37:1251-7.
22. Garand R, Avet-Loiseau H, Accard F, Moreau P, Harousseau JL, Bataille R. t(11;14) and t(4;14) translocations correlated with mature lymphoplasmacytoid and immature morphology, respectively, in multiple myeloma. Leukemia. 2003;17:2032-5.
23. Avet-Loiseau H, Garand R, Lod´e L y col. Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE, and nonsecretory multiple myeloma variants. Blood. 2003;101:1570-1.
24. Robillard N, Avet-Loiseau H, Garand R y col. CD20 is associated with a small mature plasma cell morphology and t(11;14) in multiple myeloma. Blood. 2003;102:1070-1.
25. Hundemer M, Klein U, Hose D y col. Lack of CD56 expression on myeloma cells is not a marker for poor prognosis in patients treated by high-dose chemotherapy and is associated with translocation t(11;14). Bone Marrow Transplant. 2007;40:1033-7.
26. Slomp A, Peperzak V. Role and regulation of pro-survival BCL-2 proteins in multiple myeloma. Front Oncol. 2018;8:533.
27. Touzeau C, Maciag P, Amiot M, Moreau P. Targeting Bcl-2 for the treatment of multiple myeloma. Leukemia. 2018;32:1899-907.
28. Lakshman A, Alhaj Moustafa M, Rajkumar SV y col. Natural history of t(11;14) multiple myeloma. Leukemia. 2018;32:131-8.
29. Paner A, Patel P, Dhakal B y col. The evolving role of translocation t(11;14) in the biology, prognosis, and management of multiple myeloma. Blood Rev. 2019;41:100643.
30. Chesi M, Nardini E, Lim R, Smith K, Kuehl W, Bergsagel P. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998;92:3025-34.
31. Santra M, Zhan F, Tian E, Barlogie B, Shaughnessy J Jr. A subset of multiple myeloma harboring the t(4;14)(p16;q32) translocation lacks FGFR3 expression but maintains an IGH/MMSET fusion transcript. Blood. 2003;101:2374-6.
32. Keats JJ, Reiman T, Maxwell CA y col. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood. 2003;101:1520-9.
33. Keats JJ, Maxwell CA, Taylor BJ y col. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16; q32)-positive multiple myeloma patients. Blood. 2005;105:4060-9.
34. Marango J, Shimoyama M, Nishio H y col. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood. 2008;111: 3145-54.
35. Pei H, Zhang L, Luo K y col. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470:124-8.
36. Wu SP, Pfeiffer RM, Ahn IE y col. Impact of genes highly correlated with MMSET myeloma on the survival of non-MMSET myeloma patients. Clin Cancer Res. 2016;22: 4039-44.
37. Moreau P, Attal M, Garban F y col. Heterogeneity of t(4;14) in multiple myeloma. Long-term follow-up of 100 cases treated with tandem transplantation in IFM99 trials. Leukemia. 2007;21:20204.
38. Lazareth A, Song X-Y, Coquin A y col. MB4-2 breakpoint in MMSET combined with del(17p) defines a subset of t(4;14) multiple myeloma with very poor prognosis. Haematologica. 2015;100:e471-4.
39. Du Ch, Mao X, Xu Y y col. 1q21 Gain but not t(4;14) indicates inferior outcomes in multiple myeloma treated with bortezomib. Leuk Lymphoma. 2020;61:1201-10.
40. Kataoka K, Fujiwara KT, Noda M, Nishizawa M. MafB, a new Maf family transcription activator that can associate with Maf and Fos but not with Jun. Mol Cell Biol. 1994;14:7581-91.
41. Nishizawa M, Kataoka K, Vogt PK. MafA has strong cell transforming ability but is a weak transactivator. Oncogene. 2003;22:7882-90.
42. Van Stralen E, van de Wetering M, Agnelli L, Neri A, Clevers HC, Bast BJ. Identification of primary MAFB target genes in multiple myeloma. Exp Hematol. 2009;37:78-86.
43. Hurt EM, Wiestner A, Rosenwald A y col. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. 2004;5:191-9.
44. Zhan F, Huang Y, Colla S y col. The molecular classification of multiple myeloma. Blood. 2006;108:2020-8.
45. Jenner MW, Leone PE, Walker BA y col. Gene mapping and expression analysis of 16q loss of heterozygosity identifies WWOX and CYLD as being important in determining clinical outcome in multiple myeloma. Blood. 2007;110:3291-300.
46. Walker BA, Wardell CP, Johnson DC y col. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood. 2013;121:3413-9.
47. Narita T, Inagaki A, Kobayashi T y col. t(14;16)-positive multiple myeloma shows negativity for CD56 expression and unfavorable outcome even in the era of novel drugs. Blood Cancer J. 2015;5:e285.
Clinical features and survival of multiple myeloma patients harboring t(14;16) in the era of novel agents. Blood Cancer J. 2020;10:40.
49. Ross FM, Chiecchio L, Dagrada GP y col. The t(14;20) is a poor prognostic factor in myeloma but is associated with long-term stable disease in monoclonal gammopathies of undetermined significance. Haematologica. 2010;95:1221-5.
50. Kaufmann H, Ackermann J, Baldia C y col. Both IGH translocations and chromosome 13q deletions are early events in monoclonal gammopathy of undetermined significance and do not evolve during transition to multiple myeloma. Leukemia. 2004;18:1879-82.
51. Avet-Louseau H, Daviet A, Sauner S, Bataille R. Chromosome 13 abnormalities in multiple myeloma are mostly monosomy 13. Br J Haematol. 2000;111:1116-7.
52. Shaughnessy J Jr, Tian E, Sawyer J y col. Prognostic impact of cytogenetic and interphase fluorescence in situ hybridization-defined chromosome 13 deletion in multiple myeloma: early results of total therapy II. Br J Haematol. 2003;120:44-52.
53. Binder M, Rajkumar SV, Ketterling RP y col. Prognostic implications of abnormalities of chromosome 13 and the presence of multiple cytogenetic high-risk abnormalities in newly diagnosed multiple myeloma. Blood Cancer J. 2017;7:e600.
54. Chavan SS, He J, Tytarenko R y col. Bi-allelic inactivation is more prevalent at relapse in multiple myeloma, identifying RB1 as an independent prognostic marker. Blood Cancer J. 2017;7:1-7.
55. Xiong W, Wu X, Starnes S y col. An analysis of the clinical and biologic significance of TP53 loss and the identification of potential novel transcriptional targets of TP53 in multiple myeloma. Blood. 2008;112:4235-46.
56. Jovanovic KK, Escure G, Demonchy J y col. Deregulation and targeting of TP53 pathway in multiple myeloma. Front Oncol. 2019;8:665.
57. Palumbo A, Avet-Loiseau H, Oliva S y col. Revised International Staging System for Multiple Myeloma: a report from International Myeloma Working Group. J Clin Oncol. 2015;33:2863-9.
58. Chang H, Sloan S, Li D, Keith Stewart A. Multiple myeloma involving central nervous system: high frequency of chromosome 17p13.1 (p53) deletions. Br J Haematol. 2004;127: 280-4.
59. Chang H, Qi C, Yi QL, Reece D, Keith Stewart A. p53 gene deletion detected by fluorescence in situ hybridization is an adverse prognostic factor for patients with multiple myeloma following autologous stem cell transplantation. Blood. 2005;105:358-60.
60. Tiedemann RE, Gonzalez-Paz N, Kyle RA y col. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22:1044-52.
61. Shaughnessy J. Amplification and overexpression of CKS1B at chromosome band 1q21 is associated with reduced levels of p27Kip1 and an aggressive clinical course in multiple myeloma. Hematology. 2005;10 (Suppl. 1): 117-26.
62. Hanamura I, Stewart JP, Huang Y y col. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood. 2006;108:1724-32.
63. Chang H, X Qi, A Jiang y col. 1p21 deletions are strongly associated with 1q21 gains and are an independent adverse prognostic factor for the outcome of high-dose chemotherapy in patients with multiple myeloma. Bone Marrow Transp. 2010;45:117-21.
64. Boyd KD, Ross, Walker. Mapping of chromosome 1p deletions in myeloma identifies FAM46C at 1p12 and CDKN2C at 1p32.3 as being genes in regions associated with adverse survival. Clin Cancer Res. 2011;17:7776-84.
65. Zhan F, Colla S, Wu X y col. CKS1B, over expressed in aggressive disease, regulates multiple myeloma growth and survival through SKP2- and p27Kip1- dependent and independent mechanisms. Blood. 2007;109:4995-5001.
66. Stella F, Pedrazzini E, Baialardo E, Fantl DB, Schutz N, Slavutsky I. Quantitative analysis of CKS1B mRNA expression and copy number gain in patients with plasma cell disorders. Blood Cells Mol Dis. 2014;53:110-7.
67. Fonseca R, Van Wier SA, Chng WJ y col. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia. 2006;20:2034-40.
68. Grzasko N, Hus M, Pluta A y col. Additional genetic abnormalities significantly worsen poor prognosis associated with 1q21 amplification in multiple myeloma patients. Hematol Oncol. 2013;31:41-8.
69. Sawyer JR, Tricot G, Lukacs JL y col. Genomic instability in multiple myeloma: evidence for jumping segmental duplications of chromosome arm 1q. Genes Chromosome Cancer. 2005;42:95-106.
70. Teoh PJ, An O, Chung T-H y col. Aberrant hyperediting of the myeloma transcriptome by ADAR1 confers oncogenicity and is a marker of poor prognosis. Blood. 2018;132:1304-17.
Teoh, Chung, Chng, Toh ChngIL6R-STAT3-ADAR1 (P150) interplay promotes oncogenicity in multiple myeloma with 1q21 amplification. Haematologica. 2020;105:1391-404.
Lazzari, Mondala, Delos SantosAlu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat Commun. 2017;8:1922.
73. Chapman MA, Lawrence MS, Keats JJ y col. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467-72.
74. Kulkarni MS, Daggett JL, Bender TP, Kuehl WM, Bergsagel PL, Williams ME. Frequent inactivation of the cyclin-dependent kinase inhibitor p18 by homozygous deletion in multiple myeloma cell lines: ectopic p18 expression inhibits growth and induces apoptosis. Leukemia. 2002;16:127-34.
75. Leone PE, Walker BA, Jenner MW y col. Deletions of CDKN2C in multiple myeloma: biological and clinical implications. Clin Cancer Res. 2011;14:6033-41.
76. Hebraud B, Leleu X, Lauwers-Cances V y col. Deletion of the 1p32 region is a major independent prognostic factor in young patients with myeloma: the IFM experience on 1195 patients. Leukemia. 2014;28:675-9.
77. Dickens NJ, Walker BA, Leone PE y col. Homozygous deletion mapping in myeloma samples identifies genes and an expression signature relevant to pathogenesis and outcome. Clin Cancer Res. 2010;16:1856-64.
78. Chng WJ, Huang GF, Chung TH y col. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia. 2011;25:1026-35.
79. Chiecchio L, Dagrada GP, White HE y col. Frequent upregulation of MYC in plasma cell leukemia. Genes Chromosome Cancer. 2009;48:624-36.
80. Walker BA, Wardell CP, Brioli A y col. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014;4:e191-7.
81. Møller HEH, Preiss BS, Pedersen P y col. Myc protein overexpression is a feature of progression and adverse prognosis in multiple myeloma. Eur J Haematol. 2018;101:585-90.
82. Dang CV. MYC on the path to cancer. Cell. 2012;149:22-35.
83. Dib A, Gabrea A, Glebov OK y col. Characterization of MYC translocations in multiple myeloma cell lines. J Natl Cancer Inst Monogr. 2008;39:25-31.
84. Chesi M, Bergsagel PL. Advances in the pathogenesis and diagnosis of multiple myeloma. Int J Lab Hematol .2015;37(S1):108-14.
85. Guash LG, Zurita S, Lannutti L, Pantuso F, Slavutsky I, Stella F. Evaluación de desbalances genómicos en desórdenes de células plasmáticas. Rev Inv Científicas Univ de Morón. 2020;7:33-46.
86. Rabani H, Ziv, LaviDeletions and amplifications of the IGH variable and constant regions:a novel prognostic parameter in patients with multiple myeloma. Leuk Res. 2020;99:106476.
87. Shah N, Chari A, Scott E, Mezzi K, Usmani SZ. B-cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia. 2020;34:985-1005.
88. Da Vià MC, Dietrich O, Truger M y col. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nature Med. 2021;27:616-9
89. Samur MK, Fulciniti M, Samur AA y col. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nature Comm. 2021; 12:868.
90. Stella F, Pedrazzini E, Rodríguez A y col. New recurrent chromosome alterations in patients with multiple myeloma and plasma cell leukemia. Cytogenet Genom Res. 2011;134: 249-59.
91. Walker BA, Mavrommatis K, Wardell CP y col. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33:159-70.
92. Ashby C, Tytarenko RG, Wang Y y col. Poor overall survival in hyperhaploid multiple myeloma is defined by double-hit bi-allelic inactivation of TP53. Oncotarget. 2019;10:732-7.
93. Peterson, Rowsey, MarcouHyperhaploid plasma cell myeloma characterized by poor outcome and monosomy 17 with frequently co-occurring TP53 mutations. Blood Cancer J. 2019;9:20.
94. Sawyer JR, Tian E, Shaughnessy JD Jr y col. Hyperhaploidy is a novel high- risk cytogenetic subgroup in multiple myeloma. Leukemia. 2017;31:637-44
95. Hoctor VT, Campbell LJ. Hyperhaploid plasma cell myeloma. Cancer Genet. 2012;205:414-8.
96. Mandahl N, Johansson B, Mertens F, Mitelman F. Disease-associated patterns of disomic chromosomes in hyperhaploid neoplasms. Genes Chromosome Cancer. 2012;51:536- 44.