Leucemias agudas pediátricas con alteraciones en el gen KMT2A: experiencia en los últimos 20 años en una institución
Acute pediatric leukemia with KMT2A GENE alterations: an institution experience in the last 20 years
Peruzzo L, Deu M, Pennella C, Sánchez La Rosa C, Alfaro E, Rubio P, Rossi J, Baialardo E, Guitter M, Felice M.
Hospital de Pediatría S.A.M.I.C. "Prof. Dr. Juan P. Garrahan"
luisinaperuzzo@gmail.com
Palabras claves: leucemia aguda,
pediatría,
gen KMT2A.
Keywords: acute leukemia,
pediatric,
KMT2A gene.
Introducción
La leucemia aguda (LA) es la patología oncológica más frecuente en pediatría. Las alteraciones en el gen KMT2A son detectadas en el 70% de los pacientes menores de 1 año con diagnóstico de leucemia linfoblástica aguda (LLA) y de leucemia mieloblástica aguda (LMA), en menos del 5% de niños con LLA y LMA mayores de 1 año y, aproximadamente, en la mitad de los pacientes con leucemia aguda relacionada con tratamiento oncológico previo (LART)(1). Independientemente del subtipo, las LA con reordenamientos del gen KMT2A se asocian a pronóstico adverso(2). Los objetivos del presente trabajo fueron determinar características clínicas, formas de presentación y evolución de los pacientes pediátricos con LLA, LMA y LART con alteraciones en el gen KMT2A, caracterizar subtipos y frecuencia de alteraciones que involucran al gen KMT2A y evaluar la probabilidad de sobrevida libre de eventos (SLE) y la probabilidad de sobrevida global (SG) de este grupo de pacientes.
Pacientes y métodos
Se trata de un estudio analítico, retrospectivo. Se analizaron los pacientes menores de 17 años con diagnóstico de LLA, LMA y LART con reordenamientos en el gen KMT2A diagnosticados en nuestra Institución entre enero de 2000 y julio de 2021. El compromiso del gen KMT2A fue evaluado con técnicas de reacción en cadena de polimerasa (PCR), bandeo-G y/o técnica de hibridación in situ (FISH) previamente descriptas(3,4). Se calcularon SG, SLE y error estándar (EE) y se compararon con log-rank test, con el software estadístico Graphpad Prism 5.0. Se garantizó la confidencialidad respecto a los datos personales de cada paciente (ley nacional 25.326 de Habeas Data), los cuales fueron registrados en la base datos de manera codificada. Este estudio se rigió por las normas de la Declaración de Helsinki, la Resolución Nacional Nro. 1480/2011 del Sistema de Evaluación, Registro y Fiscalización de las Investigaciones en Salud. Los protocolos de tratamiento administrados fueron aprobados por el Comité de Ética del Hospital de Pediatría Prof. Dr. Juan P. Garrahan.
Resultados
En el período estudiado se diagnosticaron 1977 pacientes con LA: 75% LLA y 25% LMA. De ellos, el 9,4% presentó reordenamientos de KMT2A, 94 LLA y 93 LMA. Con un rango etario de 0-15 años, la mediana de edad al diagnóstico fue de 6 meses para LLA y 5 años para LMA. La distribución por sexo M/F fue 1/1,22 en LLA y 1/1,44 en LMA.
La distribución según el inmunofenotipo de los blastos fue en LLA: pro-B: 46 (49%), pre-B: 34 (36,2%), linaje ambiguo: 5,4 (5%), B-madura: 3,2 (3%), B-común: 3,2 (3%), T cortical: 2 (2%) y T citotóxica: 1 (1%).
Según la clasificación FAB, la distribución de los casos de LMA fue M5: 76 (81%), LALA (mieloide y B): 4 (4%), M1:1 (1%), M2: 4 (4%), M4: 2 (2%), M7: 1 (1%), sarcoma mieloide: 3 (3%) y de estos, 10 (11%) presentaron switch de linaje: 3 LMA con switch a LLA y 7 LLA con switch a LMA.
Se diagnosticaron 5 LART secundarias a: LLA (n=3), retinoblastoma (n=1) y rabdomiosarcoma (n=1), como primera enfermedad maligna.
Se observó hepatomegalia y esplenomegalia en el 77% y 79% de los pacientes con diagnóstico de LLA y en el 60% y 45% de pacientes con LMA, respectivamente. El compromiso en piel se detectó en el 25% de los pacientes con LMA y 11% de las LLA. El SNC estaba comprometido en 15% de las LMA y 10% de las LLA y el mediastino en 10% de las LLA. Otras localizaciones comprometidas por infiltración leucémica menos frecuentes fueron riñón, páncreas y órbitas.
La mediana de recuento de leucocitos al diagnóstico fue 165.000/mm3 (rango: 2.200-1.400.000) en LLA y 16.290/mm3 (rango: 0,78-460.000) en LMA. Presentaron hiperleucocitosis (>100.000 leucocitos/mm3) el 55% de las LLA y el 37% de las LMA. Se detectaron alteraciones en el gen KMT2A por PCR en todos los casos de LLA y 80 de ellos fueron confirmados por bandeo-Gy/o por técnica de FISH (break-apart). En los casos de LMA, se caracterizaron por PCR en 65 pacientes y por bandeo-G exclusivamente en 27. La distribución de los reordenamientos del gen KMT2A en LLA fue: KMT2A-AFF1: 55%, KMT2A-MLLT1: 23,7%, KMT2A-MLLT3: 16%, KMT2A-MLLT10: 4,4%. En LMA la alteración más frecuente fue KMT2A-MLLT3 en 40% de los casos, KMT2A-MLLT10: 18%, KMT2A-MLLT11: 8%, KMT2A-AFF1 y KMT2A-MLLT1:7%. Las alteraciones menos frecuentes fueron t(X;11), KMT2A-AFDN, KMT2A-MLLT6.
Todos los pacientes recibieron tratamiento con los protocolos vigentes para LLA y LMA (Interfant, LLA-96, LLA-2002, LLA-2020 y LMA-99, LMA-08). Recibieron consolidación con trasplante de células precursoras hemopoyéticas (TCPH) 13 pacientes con diagnóstico de LLA (todos en 1ª remisión completa) y 12 con LMA (10 en 1a y 2 en 2a remisión completa).
La SLEa 5 años fue 26% (EE: 11), 43% (EE: 9) (p=0.02) y la SG 31% (EE: 10) y 47% (EE: 9) para LLA y LMA respectivamente. La SLE y SG para LART fue 25%. Se evidenciaron 69 eventos en el grupo de pacientes con KMT2A-LLA (73% del total de las LLA). De ellos fueron: 41 recaídas de enfermedad (59%), 22 muertes en remisión completa (RC) (32%), 5 muertes en inducción (7%) y un paciente tuvo respuesta nula al tratamiento. En el grupo KMT2A-LMA se evidenciaron 57 eventos (61% del total de las LMA). De éstos, 27 fueron recaídas de enfermedad (47%), 20 muertes en inducción (35%), 7 muertes en RC (13%) y 3 presentaron respuesta nula al tratamiento (5%). La SLE a 5 años en los pacientes menores y mayores de 1 años fue 22% vs 50% respectivamente (p=0.0015). Independiente del tratamiento recibido, en los pacientes menores de 1 año la SLE a 5 años en LLA y LMA fue 21% y 37% y en los menores de 6 meses 17% y 33% para LLA y LMA, respectivamente.
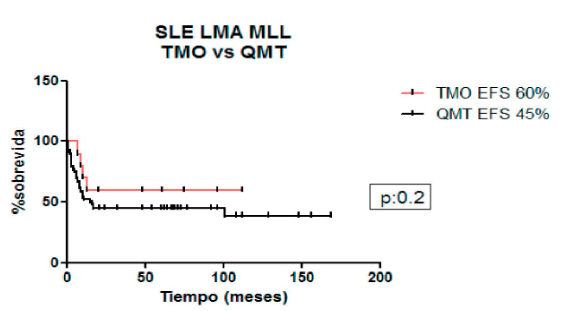
Comparando la SLE a 5 años de los pacientes que recibieron quimioterapia vs. consolidación con TCPH para la cohorte completa fue 26% vs. 50% (p=0,09), para LLA 23% vs. 46% (p=0,10) y para LMA 45% vs. 60% (p=0,20).
Figura 1. Sobrevida libre de eventos en menores y mayores de 1 año
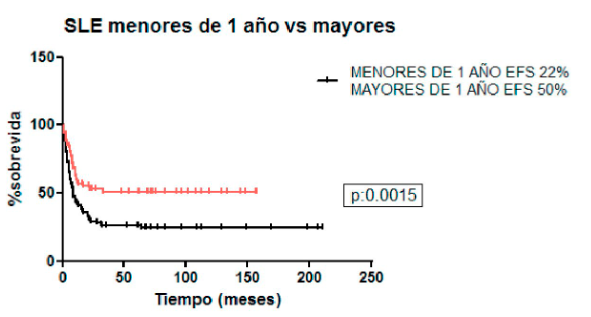
Figura 2. Sobrevida libre de eventos KMT2A LLA y KMT2A LMA
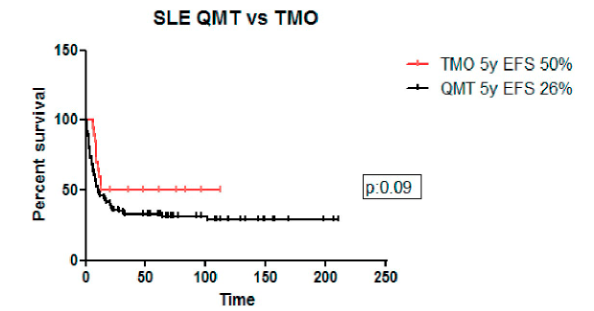
Figura 3. Sobrevida libre de eventos en pacientes con quimioterapia vs. trasplante de células precursoras hemopoyéticas
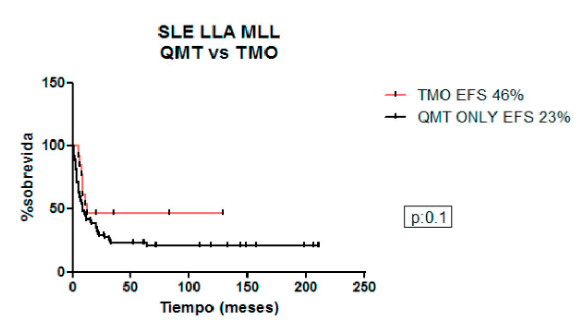
Figura 4. Sobrevida libre de eventos en pacientes con KMT2A LLA: quimioterapia vs. trasplante de células precursoras hematopoyéticas
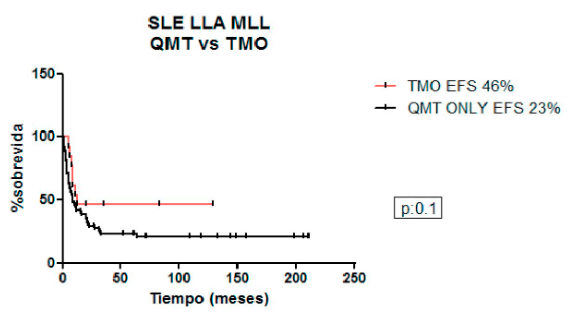
Figura 5. Sobrevida libre de eventos en pacientes con KMT2A LMA: quimioterapia vs. trasplante de células precursoras hematopoyéticas
Discusión
La LA representa una patología hematológica heterogénea caracterizada por la proliferación clonal de células hemopoyéticas inmaduras y constituye la patología maligna más frecuente en la infancia (37% del total de pacientes diagnosticados con cáncer infantil)(5). Del total de los pacientes pediátricos con diagnóstico de LA, el 75% corresponde a LLA y el restante 25% a LMA.
El gen KMT2A (Lisina metil transferasa 2A) descripto por primera vez en 1992, se encuentra localizado en el cromosoma 11q23 y codifica una proteína expresada en una amplia variedad de tejidos humanos(6). El complejo KMT2A ejerce una función epigenética crucial para el mantenimiento y desarrollo de los precursores hematopoyéticos y su diferenciación, ya que mantiene la llamada “memoria transcripcional”, lo que garantiza la identidad a las células. Lo excepcional del gen KMT2A es su gran número de posibles combinaciones con otros genes descriptos. Hasta ahora se han detectado más de 200 translocaciones en pacientes con LA, lo cual explicaría la heterogeneidad en las patologías que origina(7).
Meyer y cols. estudiaron los llamados recombinomas del gen KMT2A y describieron que los diferentes puntos de ruptura, que ocurren en el gen al translocarse, se correlacionarían con las diversas etiologías, formas de presentación clínica y evolución. Así, describieron un punto de ruptura en el intrón 11 asociado a LLA en menores de 1 año mientras los puntos de ruptura asociados a LMA o pacientes mayores se encontraba en el intrón 9(7).
Según lo reportado en la literatura, y en coincidencia con los resultados de nuestra cohorte, al analizar la frecuencia exacta de fusiones específicas de KMT2A, resulta llamativo que sólo unas pocas sean las responsables de la génesis de la mayoría de las LA. Estas fusiones son las derivadas de las translocaciones t(4;11) [KMT2A-AFF1/AF4], t(9,11) [KMT2A-MLLT3/AF9], t(10;11) [KMT2A-MLLT10/AF10] y t(11;19) [KMT2A-MLLT1/ENL] en el 92% de los pacientes con LLA, mientras que las translocaciones cromosómicas t(9;11) [KMT2A-MLLT3/AF9], t(10;11) [KMT2A-MLLT10/AF10], t(11;19) [KMT2A-MLLT1/ENL] y t(11;19) [KMT2A-ELL] son caracterizadas en el 48% de todos los pacientes con LMA(8).
Los rearreglos de KMT2A se encuentran en alrededor del 5% y 10% de los pacientes adultos y pediátricos con LA(9), respectivamente, y en la mayoría de las LA secundarias a tratamiento oncológico previo, especialmente con inhibidores de topoisomerasa II(10) confiriéndole a la enfermedad un impacto pronóstico adverso independiente(2).
Los fenotipos prevalentes en las LA con reordenamientos de KMT2A son aquéllos con características inmaduras como LLA pro-B, caracterizado por expresión de CD19, con ausencia de expresión de CD10 y frecuente co-expresión de marcadores de linaje mieloide como CD15, CD33, y CD68. Esto podría explicarse teniendo en cuenta las conclusiones de Armstrong y cols., quienes determinaron que las células con reordenamiento de KMT2A tienen un perfil de expresión génica coincidente con un estadío de desarrollo muy temprano de progenitores hematopoyéticos(11).
Los pacientes con LLA y reordenamientos de KMT2A se presentan frecuentemente con recuentos leucocitarios elevados, compromiso extramedular (nódulos subcutáneos, hepato y esplenomegalia). Erin y cols. describieron una baja frecuencia de compromiso de SNC en KMT2A-LMA (representando <10%) y alta incidencia de compromiso cutáneo como la forma de presentación más prevalente [alrededor del 80% en t(10;11)](12). En nuestra cohorte se pudo validar la alta incidencia de hiperleucocitosis como forma de presentación más frecuente en las LLA, siendo en un menor porcentaje en las LMA y la hepato y esplenomegalia como la forma de compromiso extramedular más frecuente en ambas.
La edad de presentación temprana, específicamente en menores de 1 año representa el factor pronóstico adverso más importante(13). Según lo reportado por Pui y cols., independientemente del tipo de reordenamiento de KMT2A, los niños menores de 1 año tienen peor resultado que los pacientes mayores. En esta cohorte, dos tercios de los pacientes mayores de 1 año se encontraban vivos y libres de enfermedad a los 5 años, en comparación con menos de un tercio de los pacientes menores de 1 año al momento del diagnóstico. Estos datos pudieron ser validados en una serie pequeña de pacientes con LLA de linaje B con la translocación t(11;19) donde alcanzaron la RC 5 de 7 niños de 1 a 9 años, pero sólo 2 de 13 menores de 1 año y 1 de 4 niños mayores de 10 años(14). En nuestro grupo de pacientes pudo verificarse la menor probabilidad de SLE en pacientes menores de 1 año con un valor estadísticamente significativo.
Diversos estudios intentaron demostrar, sin un alto grado de evidencia por el bajo número de pacientes, que intensificar el tratamiento con quimioterapia mejoraría los resultados de las LA que presentaban alteraciones en el gen KMT2A. En un estudio del Dana- Farber Cancer Institute Consortium, 3 de 7 pacientes menores de 1 año se encontraban vivos y libres de enfermedad a los 2, 4 y 6 años desde el diagnóstico, habiendo recibido intensificación con altas dosis de citarabina post RC(15).
La indicación de TCPH en el tratamiento de LA-KMT2A en primera RC ha sido muy controvertida a lo largo de la historia, limitándose a las sugerencias según lo reportado por grupos colaborativos internacionales. En 2002 Pui y cols. publicaron resultados de 497 pacientes pediátricos con LLA y reordenamientos de KMT2A. Todos recibieron tratamiento con quimioterapia según regímenes de alto riesgo y 56 recibieron TCPH. La SG y SLE del grupo de pacientes que recibió sólo quimioterapia fue mayor que aquéllos que se sometieron un TCPH (p=0,014) siendo la principal causa de fallo la recaída de enfermedad. El estudio tuvo como limitaciones el hecho de que los pacientes pertenecían a 12 centros, lo que determinaba la heterogeneidad de estrategias de tratamiento y que, si bien el número de pacientes fue importante, aquéllos que recibieron TCPH representaban un pequeño porcentaje(13).
En pediatría la indicación de TCPH en primera RC continúa siendo controvertida. Según los resultados del estudio Interfant 99, donde se evaluaron resultados de pacientes menores de 1 años con LLA y reordenamientos de KMT2A, no se evidenciaron beneficios con TCPH vs. tratamiento con quimioterapia, a menos que los pacientes presentaran algún factor de mal pronóstico al diagnóstico [edad menor de 6 meses, recuento de leucocitos mayor de 300.000/mm3, respuesta pobre a la prednisona o enfermedad mínima residual (EMR) detectable luego de la quimioterapia de consolidación](16).
La SG en pacientes pediátricos con LMA bajo los esquemas de tratamiento actuales es de alrededor del 60-70%, según lo publicado en la literatura(17). Generalmente estos pacientes son tratados con esquemas quimioterápicos con altas dosis de citarabina y consolidación con TCPH según el grupo de riesgo, basados en características citogenético-moleculares al diagnóstico y la respuesta al tratamiento, evaluada a través del monitoreo de la EMR(18).
En 2019 De Moerloose y cols. publicaron los resultados de un estudio multicéntrico (DB AML-01) de 112 pacientes pediátricos con diagnóstico de LMA. La sobrevida global de la cohorte fue del 74%. El 25% de los pacientes presentaba reordenamientos del gen KMT2A y, de ellos, el 14% presentaron una respuesta pobre a la terapia de inducción y el 10% fallecieron durante el primer curso de quimioterapia. Estas características establecieron que los reordenamientos de KMT2A eran determinantes de peor pronóstico de SG. Si bien no se vieron resultados favorables en aquellos pacientes que recibieron TCPH, concluyen, que aquéllos con pronóstico desfavorable serían candidatos para recibirlo en primera RC(19).
Balgobind y cols., publicaron en 2009 los resultados de un grupo amplio de pacientes pediátricos con LMA y reordenamientos de KMT2A. Todos los pacientes recibieron terapia de inducción con antraciclínicos y altas dosis de citarabina, y de los 756 pacientes totales, 108 recibieron TCPH. En el análisis de multivarianza el TCPH no demostró ser un predictor de mejor SLE y SG, otro trabajo limitado por el escaso número de pacientes que recibieron TCPH comparado con el número total de la cohorte(20).
Según los resultados arrojados en un estudio comandado por el Children's Oncology Group (Trial AAML0531) en pacientes con LMA y reordenamientos de KMT2A(12) la presencia de t(6;11)(q27;q23), t(10;11)(p11.2;q23) y t(10;11)(p12;q23) fueron predictores desfavorables independientes de inferior SG y SLE. Debido a que presentan un alto riesgo de recaída, debería considerarse el TCPH en primera RC.
En coincidencia con lo reportado en la literatura, en nuestro grupo de pacientes pudimos concluir que los pacientes con reordenamientos de KMT2A representan un grupo de alto riesgo, tanto en LLA como LMA, pudiendo beneficiarse con TCPH en primera RC, con el objetivo de prevenir la recaída de la enfermedad, que representa la principal causa de fallo al tratamiento y una pobre SG, más allá de las características desfavorables de presentación que contribuyen a la evolución tórpida.
Conclusiones
Las LA con alteraciones en el gen KMT2A corresponden a un grupo especial con pobre pronóstico, independientemente del subtipo génico involucrado. Los pacientes menores de 1 año y, en especial, los menores de 6 meses, presentaron las tasas de sobrevida más pobres. El grupo de pacientes que recibió TCPH mostró una tendencia a presentar mejores resultados de sobrevida tanto en LLA como en LMA.
Bibliografía
1. Daser A, Rabbitts TH. The versatile mixed lineage leukaemia gene MLL and its many associations in leukaemogenesis. Semin Cancer Biol. 2005 Jun; 15(3):175-88.
2. Balgobind BV, Raimondi SC, Harbott J et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 114,2489-2496.
3. Gabert J, Beillard E, van der Velden VH et al. Standardization and quality control studies of 'real-time' quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia - a Europe Against Cancer program. Leukemia. 2003 Dec;17(12):2318-57.
4. Shaffer LG, McGowan-Jordan J, Schmid. ISCN: an International System for Human Cytogenetic Nomenclature. Basel (Switzerland): S. Karger AG.
5. Moreno Florencia; Registro Oncopediátrico Hospitalario Argentino 7ma. Edición. Tendencia de Incidencia 2000-2019, Supervivencia 2005-2014.
6. Butler LH, Slany R, Cui X et al.. The HRX proto-oncogene product is widely expressed in human tissues and localizes to nuclear structures. Blood. 1997;89(9):3361-70.
7. Meyer C, Burmeister T, Gröger D et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018 Feb;32(2):273-284.
8. Marschalek R. MLL leukemia and future treatment strategies. Arch Pharm (Weinheim). 2015 Apr;348(4):221-8.
9. Moorman AV, Ensor HM, Richards SM et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11(5):429-38.
10. Andersen MK, Christiansen DH, Jensen BA et al. Therapy-related acute lymphoblastic leukaemia with MLL rearrangements following DNA topoisomerase II inhibitors, an increasing problem: report on two new cases and review of the literature since 1992. British Journal of Haematology. 2001;114:539-543.
11. Armstrong SA, Staunton JE, Silverman LB et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2001;30: 41-47.
12. Erin M. Guest, Betsy A. Hirsch, E. Anders Kolb et al. Prognostic Significance of 11q23/MLL Fusion Partners in Children with Acute Myeloid Leukemia (AML) - Results from the Children's Oncology Group (COG) Trial AAML0531. Blood. 2016;128 (22):1211.
13. Pui CH, Gaynon PS, Boyett JM et al. Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet. 2002 Jun 1;359(9321):1909-15.
14. Rubnitz JE, Camitta BM, Mahmoud H et al. Childhood acute lymphoblastic leukemia with the MLL-ENL fusion and t(11;19)(q23;p13.3) translocation. J Clin Oncol. 1999; 17: 191-96.
15. Silverman LB, McLean TW, Gelber RD et al. Intensified therapy for infants with acute lymphoblastic leukemia: results from the Dana- Farber Cancer Institute Consortium. Cancer. 1997;80:2285-95.
16. Mann G, Attarbaschi A, Schrappe M et al. Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage leukemia (MLL)-rearranged acute lymphoblastic leukemia: results from the Interfant-99 Study. Blood. 2010;116(15):2644-50.
17. Abrahamsson J, Forestier E, Heldrup J et al. Response-guided induction therapy in pediatric acute myeloid leukemia with excellent remission rate. JCO. 2011;29:310-315.
18. von Neuhoff C, Reinhardt D, Sander A et al. Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AMLBFM 98. JCO. 2010;28:2682-2689
19. De Moerloose, B, Reedijk, A, de Bock, GH et al. Response-guided chemotherapy for pediatric acute myeloid leukemia without hematopoietic stem cell transplantation in first complete remission: Results from protocol DB AML-01. Pediatr Blood Cancer. 2019; 66:e2760.
20. Balgobind B, Raimondi S, Harbott J et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study Blood. 2009;114(12):2489-2496.