Aplasia eritrocítica pura y síndrome antifosfolípido primario: una asociación autoinmune
Pure red cell aplasia and primary antiphospholipid syndrome: an autoimmune association
Ghayad E; Kadri Z, Haddad F
Departamento de Medicina Interna e Inmunología Clínica, Hospital Hôtel-Dieu de France, Universidad Saint-Joseph, Beirut - Líbano.
elieghayad@gmail.com
Palabras claves: anemia,
antifosfolípido,
autoinmunidad.
Keywords: anemia,
antiphospholipid,
auto-immunity.
Resumen
La aplasia eritrocítica pura es una causa poco frecuente de anemia no regenerativa debida a una supresión autoinmune de la eritropoyesis. Se ha asociado clásicamente con el lupus eritematoso sistémico y la artritis reumatoidea. Presentamos el caso de un paciente con síndrome antifosfolípido primario que desarrolló una anemia grave concordante con una aplasia eritrocítica pura. Esta asociación refuerza la base autoinmune de la aplasia eritrocítica pura.
Abstract
Pure red cell aplasia is an uncommon cause of non-regenerative anemia due to an autoimmune suppression of erythropoiesis. It has been classically associated with systemic lupus erythematosus and rheumatoid arthritis. We report the case of a patient with primary antiphospholipid syndrome who developed severe anemia concordant with pure red cell aplasia. The aforementioned association reinforces the autoimmune basis of pure red cell aplasia.
Introduction
Pure red cell aplasia (PRCA) is a rare cause of anemia. This diagnosis should be considered by the clinician investigating an anemia with a negative initial workup, particularly if an associated medical condition is present.
Case description
We report the case of a 34-year-old patient with a 10-year history of antiphospholipid syndrome diagnosed after an anterior acute coronary syndrome secondary to an ostial left anterior descending (LAD) coronary artery stenosis which required coronary artery bypass grafting.
Serological studies were positive for lupus anticoagulant, anticardiolipin antibodies (IgM & IgG) and anti-beta-2-glycoprotein antibodies confirmed on multiple testing. Of note antinuclear antibodies were negative.
He also developed progressive peripheral artery disease with intermittent claudication requiring a right femoral popliteal bypass surgery.
He was treated with warfarin adjusted to an INR target of 2-3, nebivolol, sulodexide, irbesartan and atorvastatin.
The patient was admitted in Hôtel-Dieu de France hospital, Beirut, Lebanon for acute chest pain radiating to the left arm associated with normal electrocardiogram, elevated cardiac markers, and anemia. Coronary angiography revealed a new 50% LAD stenosis along with patent coronary bypass grafts: the chest pain was then attributed to a type 2 myocardial infarction.
At admission the patient had an anemia characterized by a hemoglobin level of 8 g/dL (hematocrit level of 25%) with a normocytic normochromic profile and a notably very low reticulocyte count (5440/mm3; 0.1% corrected for hematocrit level). Iron studies revealed normal serum iron with high ferritin level, and elsewhere the patient had normal vitamin B9, B12 and TSH measurements. Concerning hemolysis biomarkers, haptoglobin and unconjugated bilirubin dosages were within the reference range not indicating hemolysis along with a mildly elevated lactate dehydrogenase level; direct and indirect Coombs testing were negative. Elsewhere, peripheral blood smear showed anisocytosis and poikilocytosis.
Other blood examinations done on admission revealed a normal kidney and liver function tests.
Multiple packed red blood cell transfusions were administered with improvement in the patient’s symptoms.
A bone marrow aspirate and biopsy were done and were compatible with pure red cell aplasia, showing a normocellular marrow with absence of erythroid precursors.
Figure 1. May Grunwald Giemsa staining of bone marrow aspirate showing a remarkable absence of erythroblasts.
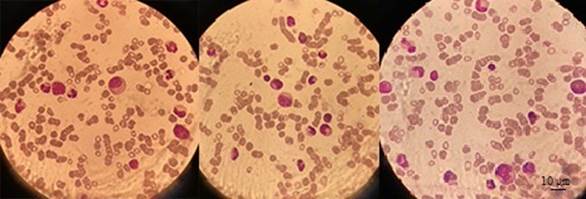
Etiological differential diagnoses were eliminated: thymoma was ruled out by thoracic computed tomography; lymphoma and myelodysplastic syndrome were also ruled out by the bone marrow studies; serologies for parvovirus B19, hepatitis B & C and human immunodeficiency virus were negative as well as plasma polymerase chain reaction for cytomegalovirus and Epstein-Barr virus; drug history did not reveal any possibly causative agent. Complement levels were normal and antinuclear antibodies were negative. Paroxysmal nocturnal hemoglobinuria was eliminated on the basis of the absence of hemolysis or other cytopenias; flow cytometry was not performed.
The patient was subsequently treated with methylprednisolone intravenous pulse therapy for 5 days then discharged on prednisone 1 mg/kg/day with subsequent tapering and cyclosporin 3 mg/kg/day divided in two doses with blood testing follow-up. Of note hemoglobin measurement on the final day of hospitalization was 7.7 g/dL.
On follow-up after two months, the patient was asymptomatic, with blood tests showing a hemoglobin level of 10 g/dL (hematocrit 29.7%) and a corrected reticulocyte count of 1%, marking an improvement of his condition. Cyclosporin treatment was maintained, and prednisone tapering continued at a rate of 10 mg per week until 20 mg daily. Antiphospholipid antibodies dosing was planned for the next follow-up appointment.
Discussion
Pure red cell aplasia is a syndrome defined by a normocytic normochromic anemia with severe reticulocytopenia and marked reduction or absence of erythroid precursors from the bone marrow (1), without affecting other lineages. PRCA may be either congenital (Diamond-Blackfan anemia) or acquired and if the latter is the case, primary PRCA should be distinguished from PRCA secondary to other etiologies, such as infectious, neoplastic (lymphoproliferative disorders, hematologic malignancies, solid tumors) or auto-immune/collagen vascular diseases(1).
Concerning auto-immune diseases, PRCA has been particularly described in association with systemic lupus erythematosus(2) and rheumatoid arthritis(3). In some cases, patient serum may inhibit the growth of erythroid precursor cells of both the patient and control, while in other cases the inhibition may be mediated by T-lymphocytes(4).
In previous reports, Walton et al first described a case of PRCA and antiphospholipid syndrome (APS) in a female patient who also presented with hemolytic anemia and had positive anti-DsDNA antibodies compatible with systemic lupus erythematosus(5). The case of a patient with APS who developed PRCA and neuro-lupus was also reported by Murayama et al(6).
To our knowledge the association between PRCA and APS without concurrent lupus erythematosus has been described once in the medical literature, by Caldas et al(7). In this case, the patient had been taking chlorpromazine for 13 years, a drug known for its myelotoxicity(8) and for its ability to induce the production of antiphospholipid antibodies(9). As noted by the authors(7), the anticardiolipin antibodies persisted at high titer after drug discontinuation.
We present the case of a patient known to have primary APS with negative anti-nuclear antibodies who developed PRCA, with no other concurrent contributing factor. Antiphospholipid antibodies can cross-react with erythrocyte membranes, as shown by Del Papa et al, with antibodies eluted from red blood cells displaying anti-cardiolipin binding activity(10).
In regard to management, we treated the patient as per the standard of care for primary PRCA with corticosteroids and cyclosporine(1).
In conclusion, antiphospholipid syndrome should be considered in the differential diagnosis for pure red cell aplasia in the appropriate clinical setting.
The patient gave informed consent for the publication of his/her case.
The authors declare that there is no conflict of interest.
References
1. Means RT Jr. Pure red cell aplasia. Hematol Am Soc Hematol Educ Program. 2016 Dec 2;2016(1):51-6.
2. Lobbes H, Mahevas M, Alviset S, Galicier L, Costedoat-Chalumeau N, Amoura Z et al. Pure Red Cell Aplasia in Systemic Lupus Erythematosus, a Nationwide Retrospective Cohort and Review of the Literature. Blood. 2020 Nov 5;136(Supplement 1):45-6.
3. Dessypris EN. Rheumatoid Arthritis and Pure Red Cell Aplasia. Ann Intern Med. 1984 Feb 1;100(2):202.
4. Dessypris EN. The biology of pure red cell aplasia. Semin Hematol. 1991;28(4):275-84.
5. Walton T, Karim Y, Wright D, Khamashta M, Hughes G. The association of pure red cell aplasia with the antiphospholipid syndrome. Lupus. 2001 Dec;10(12):899-901.
6. Murayama J, Asanuma Y, Tsuda T, Nishida J, Moriguchi M. Appearance of central nervous system lupus during corticosteroid therapy and warfarinization in a patient with pure red cell aplasia and antiphospholipid syndrome. Jpn J Clin Immunol. 2006;29(1):43-7.
7. Caldas CAM, de Carvalho JF. Pure red cell aplasia and primary antiphospholipid syndrome: a unique association. Rheumatol Int. 2012 May;32(5):1363-5.
8. Benestad HB. Aplastic anaemia: considerations on the pathogenesis. Acta Med Scand. 2009 Apr 24;196(1-6):255-62.
9. Mintzer DM, Billet SN, Chmielewski L. Drug-Induced Hematologic Syndromes. Matutes EM, editor. Adv Hematol. 2009 Jul 7;2009:495863.
10. Del Papa N, Meroni PL, Barcellini W, Borghi MO, Fain C, Khamashta M, et al. Antiphospholipid antibodies cross-reacting with erythrocyte membranes. A case report. Clin Exp Rheumatol. 1992 Aug;10(4):395-9.