Sarcoma histiocítico en paciente con antecedente de leucemia linfoblástica aguda. Reporte de un caso
Histiocytic sarcoma in patient with previously acute lymphoblastic leukemia. Case report
Morici M1, León Rivadeneira C1, Deana A1, Sala J1, Marchisella M1, Cafferata C2, Zamora V2, Galluzzo L3, Reichel P2, Noriega S1, Riccheri MC1
1. Hospital Nacional Profesor Dr. A. Posadas
2. Hospital CePSI Eva Perón (SDE),
3. Hospital de Pediatría J. P. Garrahan.
mercedesmorici@gmail.com
Palabras claves: sarcoma histiocítico,
pediátrico,
tratamiento.
Keywords: histiocytic sarcoma,
pediatric,
treatment.
Resumen
El sarcoma histiocítico (SH) es una neoplasia maligna poco frecuente, con características morfológicas e inmunofenotípicas semejantes a los histiocitos maduros, que representa menos del 1% de todas las neoplasias malignas hematológicas. El SH puede asociarse a linfomas, mielodisplasia y algunas leucemias. Su presentación clínica varía desde ser localizado hasta presentar compromiso sistémico. Se presenta el caso clínico de un varón de 11 años de edad, con antecedente a los 6 años de leucemia linfoblástica aguda B, en remisión continua completa (RCC). A los 2 años de finalizado su tratamiento presenta un cuadro clínico de dolor abdominal y vómitos con fiebre intermitente y TAC abomino-pélvica con masa expansiva a nivel mesentérico. Con diagnóstico anatomopatológico de sarcoma histiocítico realizan 2 ciclos de quimioterapia preoperatoria, posteriormente hemicolectomía derecha con resección completa. Por márgenes microscópicos comprometidos y ganglios infiltrados se decide continuar quimioterapia, completando dos ciclos más. A los 7 meses del diagnóstico se constata por PET-TC 2 adenopatías retroperitoneales metabólicamente activas, que se resecan completamente. En control PET-TC a los 9 meses post última cirugía no se evidencian adenopatías hipercaptantes. Actualmente permanece en RCC a 2 años del diagnóstico.
AbstractHistiocytic sarcoma (HS) is a rare malignant neoplasm, with morphological and immunophenotypic characteristics similar to mature histiocytes, representing less than 1% of all hematological malignancies. HS can be associated with lymphomas, myelodysplasia, and some leukemias. Its clinical presentation varies from localized to presenting systemic compromise. We present the clinical case of an 11-year-old boy with a 6-year old history of acute lymphoblastic leukemia B, in complete continuous remission (CCR). Two years after finishing his treatment, the patient presents symptoms of abdominal pain and vomiting, intermittent fever and an abomino-pelvic CT with an expansive mass at the mesenteric level. With an anatomopathological diagnosis by biopsy of histiocytic sarcoma, he performed 2 cycles of preoperative chemotherapy, followed by a right hemicolectomy with complete resection. Due to compromised microscopic margins and infiltrated lymph nodes, it was decided to continue chemotherapy, completing two more cycles. Seven months after the diagnosis, 2 metabolically active retroperitoneal lymphadenopathies were confirmed by PET-CT which were completely resected. With PET-CT control 9 months after the last surgery, there was no evidence of hyper-uptake lymphadenopathy. He currently remains in CCR 2 years after diagnosis.
Introducción
El sarcoma histiocítico (SH) es una neoplasia maligna poco frecuente, que representa menos del 1% de todas las neoplasias malignas hematológicas. Puede afectar ganglios linfáticos o sitios extraganglionares como piel, aparato gastrointestinal o tejidos blandos. Y se suele presentar como enfermedad avanzada y con un curso clínico agresivo.
La última clasificación de la OMS (2016) define el SH como una neoplasia maligna con características morfológicas e inmunofenotípicas semejantes a los histiocitos maduros. El SH puede asociarse a linfomas, mielodisplasia y algunas leucemias. El diagnóstico se basa en la verificación del linaje histiocítico (inmunohistoquímica) y la exclusión de otras neoplasias malignas de células grandes poco diferenciadas.
La edad de presentación más frecuente es en adultos, con leve predominio de género masculino. Las manifestaciones clínicas varían según el sitio afectado, pudiendo ser localizado o con compromiso sistémico como fiebre, anorexia, astenia, mal estado general, pérdida de peso, pancitopenia y hepatoesplenomegalia. Cuando la localización es gastrointestinal suele manifestarse con dolor abdominal, distensión, y hasta signos de oclusión intestinal.
Caso clínico
Paciente de 11 años de edad, sexo masculino, con antecedente de leucemia linfoblástica aguda B a los 6 años de vida. Realizó tratamiento según protocolo GATLA ALLIC BFM 2010, para riesgo intermedio, alcanzando remisión completa al día 33. Finalizó mantenimiento en agosto de 2017.
En mayo de 2019, consultó por dolor abdominal, vómitos de una semana de evolución y fiebre intermitente. Se realizó tomografía computada de abdomen donde se identificó a nivel mesentérico una masa inframesocólica derecha, lobulada, de contornos regulares que realzaba con contraste EV, de 141 x 99 x 133 mm (volumen tumoral: 965 cc). Resto del estudio sin particularidades (Figura 1).
Figura 1
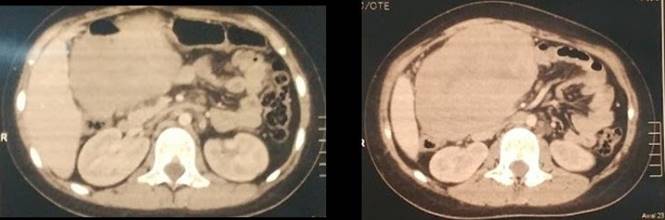
Se completaron estudios con centellograma óseo normal y biopsia de médula ósea sin enfermedad.
Se realizó cirugía con toma de biopsia del tumor abdominal donde se constató sarcoma histiocítico.
Informe anatomopatológico
Macroscopia: tumoración esferiforme de tejido de 11 x 11 x 6,4 cm que pesa 655 g. Presenta superficie externa multinodular, brillante, rosado-pardusca con focos de congestión y hemorragia. Consistencia firme elástica, bien delimitada, sólida, heterogénea, que alterna áreas blanquecinas con otras parduscas y sectores friables naranja-amarillentos que representa el 60 % de la lesión (Figura 2).
Figura 2

Microscopía: proliferación neoplásica constituida por células pleomórficas de gran tamaño, con núcleos vesiculosos con plegamientos de la membrana, cromatina granular fina y presencia ocasional de nucléolo evidente. Abundante citoplasma eosinófilo o claro, con fagocitosis de células plasmáticas o polimorfonucleares. Se evidencian células con multinucleación y formas bizarras. Alto índice mitótico. La proliferación se dispone en forma discohesiva con un patrón de crecimiento sólido con tractos fibrosos gruesos y delimitada por una gruesa cápsula fibrosa. Presenta necrosis y hemorragia tumoral en aproximadamente 12 % de la superficie evaluada (Figura 3).
Inmunohistoquímica: CD68, S100 y lisozima: positivos. CD1a, CD2, CD23, CD3, CD8, MPO, CK, CD19, CD20, CD21, CD34; HMB45, CD79a negativos. BRAF negativo, Ki 67: 15 % aproximadamente (Figuras 4 y 5).
Figura 3

Figura 4

s100
Figura 5

Lisozima CD68
Diagnóstico: sarcoma histiocítico. Rango mitótico: Puntaje 1. Necrosis: aproximado 12 % corresponde a puntaje 1 (menos del 50 %). Grado histológico: 2. Márgenes: libre de infiltración, el más cercano a 0.1 cm (bloque 5i). Invasión linfo-vascular: presente. Nódulo linfático regional: Nódulo linfático regional: compromiso de dos ganglios linfáticos (2/5). Estadificación patológica: pT3 (tumor con invasión a otros órganos: serosa colónica). pN1.
Realizó dos ciclos de CHOPP (ciclofosfamida, meprednisona, vincristina y doxorrubicina) con franca reducción clínica de la masa abdominal.
Posteriormente resección quirúrgica macroscópicamente completa de la masa, y hemicolectomía derecha con anastomosis ileo colónica.
Por márgenes microscópicos comprometidos y ganglios infiltrados se decidió continuar quimioterapia, completando dos ciclos más de CHOPP post quirúrgicos. Continuó con controles por imágenes.
En PET-TC (7 meses del diagnóstico) se observaron dos adenopatías retroperitoneales metabólicamente activas a nivel de retroperitoneo intercavo aórtico, una de 10 x 16 mm y otra de 7 x 11 mm, SUV 8 y 22 respectivamente.
Se realizó RMN que evidenció imagen nodular a nivel interaórtico de 33 x 38 mm con realce heterogéneo tras el contraste.
Se realizó nueva resección quirúrgica de masa tumoral y colocación de prótesis aórtica entre la emergencia de la arteria mesentérica inferior y arterias renales.
Se repite PET-TC post quirúrgico no evidenciándose actividad metabólica (Figura 6).
Figura 6

Actualmente el paciente se encuentra a 17 meses de la última intervención quirúrgica, sin evidencia de enfermedad.
Discusión
El SH es una neoplasia maligna extremadamente rara. Con características morfológicas e inmunofenotípicas de histiocitos, se clasifica dentro de los tumores del linaje de células dendríticas-macrófagos(1,2).
El SH se presenta principalmente en adultos con leve predominio masculino. Involucra principalmente sitios extraganglionares como la piel, tejidos blandos superficiales y profundos, tracto gastrointestinal y sistema hematopoyético(3).
Se describen presentaciones clínicas variables, que van desde enfermedad localizada con una masa solitaria a diseminación sistémica.Algunos pacientes tienen antecedentes de trastornos hematolinfoides como linfoma folicular, leucemia linfocítica crónica, linfoma de zona marginal, linfoma de células del manto, leucemias linfoblástica aguda y mieloma múltiple. También puede haber asociación con tumor de células germinales en el adulto y síndrome linfoproliferativo autoinmune en pediatría(1).
El sarcoma histiocítico es a menudo muy agresivo, con una mediana de supervivencia de varios meses en pacientes con enfermedad diseminada, mientras que los pacientes con enfermedad localizada pueden sobrevivir años después del diagnóstico.Macroscópicamente se presenta como una masa de límites delimitados o infiltrativos, con hemorragia y necrosis variable. Histológicamente se trata de una proliferación neoplásica constituida por células pleomórficas de tamaño mediano y grande, con núcleos vesiculosos y plegamientos de la membrana nuclear, cromatina granular fina y presencia ocasional de nucléolo. La actividad mitótica, la necrosis tumoral y el infiltrado inflamatorio de fondo mixto suelen ser prominentes.La inmunohistoquímica es fundamental en el diagnóstico del SH para confirmar la estirpe macrofágica y descartar otras neoplasias de células grandes y epitelioides que puedan imitarlo.La mayoría de los SH expresan CD68, CD163 y PU1, con un subconjunto de casos que también expresan CD31, CD4 (citoplasmático), CD45RO y CD15(2,3), aunque el criterio diagnóstico exige la expresión de al menos 2 de los siguientes marcadores: CD68, CD163, CD4 y lisozima, además de la negatividad para células mieloides (CD13, MPO), sobre todo en pediatría. Con el mismo criterio la proliferación debe ser negativa para células de Langerhans (CD1a, langerina). En la población general se debe considerar excluir la diferenciación melanocítica (SOX10, HMB-45, MART-1), epitelial (queratina, EMA), vascular (ERG), marcadores específicos de células B y células T (CD20, PAX5, CD3) y también marcadores de células dendríticas foliculares (CD21, CD35)(2,5).
El diagnósticos diferencial del SH es amplio, incluye al linfoma anaplásico de células grandes, linfoma no Hodgkin de células B, linfoma de Hodgkin, sarcomas pleomórficos no clasificados y tumores células dendríticas(1,4).
Se deben considerar otras enfermedades histiocíticas, como la histiocitosis de células de Langerhans y el sarcoma de células de Langerhans. Melanoma, carcinoma metastásico y rabdomiosarcoma pleomórfico son entidades a plantearse desde la morfología en el espectro de pacientes adultos.Se ha sugerido que el SH surge de la transdiferenciación de neoplasias hematolinfoides preexistentes, por lo que podría compartir alteraciones moleculares con dichas enfermedades. Se encontró la fusión H (IgH) - BCL2 en pacientes con SH y linfoma folicular. La fusión ciclina D1-IgH ha sido reportada en el SH así como en el linfoma de células del manto. La mutación V600E de BRAF ha sido también identificada. Las mutaciones BRAF pueden actuar como mutaciones impulsoras que favorecen las transformaciones malignas en los trastornos histiocíticos a través de la activación constitutiva de la vía RAS / RAF. Se han identificado mutaciones recurrentes en la vía MAP quinasa, que incluye KRAS, NRAS y MAP2K1, junto con mutaciones en KMT2D y ARID1A(5,7).
El SH sistémico tiene un pronóstico agresivo y la supervivencia general no se extiende más allá de los dos años.No existe un protocolo estándar para el manejo del SH debido a la rareza de la enfermedad. Se describen diferentes regímenes, incluidos CHOP, CHOP-E, BEAM y MEAM.La terapia más popular para la enfermedad avanzada es el régimen de ciclofosfamida, doxorrubicina, vincristina, prednisona (CHOP) con el agregado de etopósido al régimen CHOP.También están descriptos la talidomida, alemtuzumab y vemurafenib en pocos casos individuales con buenas respuestas terapéuticas(7).
El alo-trasplante de progenitores hematopoyéticos se reserva principalmente para SH recidivante y se han documentado pocos casos con respuesta completa(8).
El SH localizado requiere cirugía. En algunos casos está descripta la radioterapia.
Conclusión
Es importante tener presente esta entidad como diagnóstico diferencial principalmente de linfomas y sarcomas, sobre todo en pacientes con antecedentes de enfermedad linfoproliferativa.
Las técnicas de inmunohistoquímica son fundamentales para la confirmación del diagnóstico.
Debido a la rareza de esta entidad, la bibliografía al respecto, en su mayoría, son reportes de casos.
No existe un consenso sobre el manejo más adecuado de esta patología. La resección quirúrgica completa es el tratamiento de elección para la enfermedad localizada, mientras que para la sistémica se describe quimioterapia, radioterapia y en algunos casos alo-trasplante de progenitores hematopoyéticos.
El pronóstico es desfavorable en la enfermedad sistémica, con alta tasa de recidivas y alta mortalidad.
Bibliografía
1. Hung YP, Qian X. Histiocytic Sarcoma.Arch Pathol Lab Med. 2020 May; 144(5):650-654.
2. Hornick JL, Jaffe ES, Fletcher Ch. Extranodal histiocytic sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am J Surg Pathol. 2004 Sep; 28 (9):1133-44.
3. Kommalapati A, Harsha Tella S. Predictors of survival, treatment patterns, and outcomes in histiocytic sarcoma. Leukemia & Lymphoma. 2019 Feb;60(2):553-555.
4. Tocut M, Vaknine H, Potachenko P y col. Histiocytic Sarcoma. Med Assoc J. 2020 Oct; 22(10):645-647.
5. Hung YP, Lovitch SB, Qian X. Histiocytic sarcoma: New insights into FNA cytomorphology and molecular characteristics. Cancer Cytopathol. 2017 Aug; 125(8):604-614.
6. Ansari J, Naqash A, Munker R y col. Histiocytic Sarcoma as a Secondary Malignancy: Pathobiology, Diagnosis, and Treatment. Eur J Haematol. 2016 Jul; 97 (1):9-16.
7. Go H, Jeon YK, Huh J y col. Frequent detection of BRAF (V600E) mutations in histiocytic and dendritic cell neoplasms. Histopathology. 2014 Aug; 65(2):261-72.
8. Abu-Sanad A, Warsi A, Michel RP, Nahal A y col. Long-term remission after autologous stem-cell transplantation for relapsed histiocytic sarcoma. Curr Oncol. 2012; 19:e289-91.